









 您已经拒绝加入团体
您已经拒绝加入团体

 2025-08-06
2025-08-06
 6705
6705
 0
0
【摘要】 深入解读《模拟计算指南》4.3节"芳香性":涵盖能量尺度、几何结构、磁学性质等多维分析方法,详解NICS、AICD等前沿技术,附专家力荐。本书由唯理计算团队历时7年打造,提供计算化学从理论到实战的全流程指导,点击了解书籍及计算服务。

✎ 关于本书
《模拟计算指南》是唯理计算工程师团队沉淀7年实战经验、历时一年打造,是一本计算化学快速入门指南、材料模拟计算领域的实用宝典。
“书中详细介绍了从理论计算化学的基本原理到目前国际前沿应用体系的计算模拟思路和方法,有利于读者从多维度理解如何采用理论计算方法来解决复杂科学问题,并帮助初学者从中找到适合自己科研的理论支持和计算解决方案。”
——教育部长江学者、杰青、复旦大学教授
刘智攀
“本书以其实用性和易学性为特色,无论是计算物质科学的初学者还是资深研究者,都能从中获得独特的视角和丰富的知识资源,使其成为该领域内一本极具价值的入门及参考书籍。”
——教育部长江学者特聘教授、华南师范大学教授
赵纪军

↑扫码了解更多书籍及唯理计算信息
01文章介绍
今天我们介绍下《模拟计算指南》的4.3 芳香性。
芳香性是一个古老又现代、基础又复杂的概念。最早人们发现煤焦油中蒸馏出来的部分碳氢化合物有独特的气味,故将其称为芳香化合物。又随着它们独特反应性的陆续发现,而将它们的性质冠以了芳香性的名字。时至今日,芳香性已经成为涵盖能量、几何结构、磁学性质、光电性质、电子结构等诸多方面的多维度的概念。这些维度上的性质可能均表现出芳香性,也可能只在部分维度上表现出芳香性。芳香性的概念早已超越了最早平面共轭分子的限制,衍生出Baird芳香性、Mobius芳香性、金属芳香性、三维芳香性等诸多蓬勃发展的新领域。
芳香性最原始的含义来自能量尺度。与相同大小的链状共轭烯烃相比,以苯为代表的特定环状共轭芳烃表现出额外的稳定性,例如格外难以被氢化、格外低的燃烧热等。人们将其归纳为“芳香稳定化能”,即由于满足特定的条件而带来的、与同类非芳香的共轭多烯相比的额外稳定化能,并且设计了许多方法来从热力学上探讨芳香性带来的能量影响,将其与其他因素剥离开。例如,考察如图4.11所示的等键反应的焓变,并将其作为芳香性稳定化能。
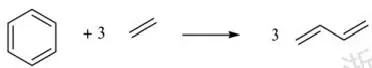
图4.11 通过乙烯和丁二烯定义的用于衡量苯芳香性的等键反应
在这个反应中,左右两侧都有12个sp²杂化的碳原子,可以视为苯中的环状共轭体系向丁二烯中的开链共轭体系转化的焓变,一定程度上可以反映苯因环状共轭结构而额外带来的稳定化能。
显然,能量尺度的性质非常容易测量和计算。只要对苯、乙烯、丁二烯进行构型优化和频率计算,即可得到上述定义下的芳香稳定化能。这种做法的局限性也显而易见,它高度依赖于设计合适的等键反应或热力学循环,而对于稍微复杂的体系,这都是不现实的。因此上述能量角度虽然在历史上对于建立芳香性概念起到了重要作用,但现实研究中很难加以考察。
芳香性分子在几何结构上往往有典型的特征。与普通共轭体系相比,芳香性体系呈现出更加明显的键长平均化和分子平面化,因此通过考察几何结构特别是键长,可以对芳香性带来直观的认识。以M06-2X/def2-TZVP水平下环丁二烯和苯的结构为例(图4.12),可以明显发现两者键长交替的情况大有不同,前者为反芳香性,后者为芳香性。
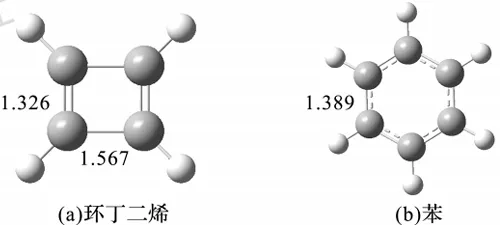
图4.12 环丁二烯和苯的几何结构
通过HOMA(harmonic oscillator measure of aromaticity)方法,可以将键长平均化的程度定量化。HOMA的定义如下:
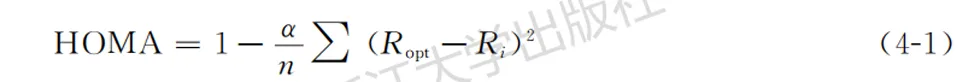
其中,Ri为某类化学键的实际键长;Ropt、α均为原作者对同类化学键确定好的固定数值;n为该化学键的个数。HOMA只依赖于几何结构,只需将不论任何手段得到的键长代入即可;数值越偏离1,意味着键长交替的程度越大,即芳香性越弱。HOMA的优点是方便快捷。其缺点也很明显:其一原作者只给少数化学键确定了参数;其二它只反映几何结构的信息,无法包含芳香性的诸多内涵。
现代对芳香性的最主要认识来自磁学性质。芳香化合物在核磁共振谱图中呈现鲜明的屏蔽区和去屏蔽区,很容易识别出来,因此对核磁的研究成为人们获得对芳香性认识的重要手段。显然,通过对化合物中芳环周边氢原子核磁化学位移的计算,可以获得对芳香性的认识。然而,特定原子的化学位移容易受到除芳香性之外因素的干扰;为了将芳香性的影响尽可能分离出来,人们发展出了核独立化学位移(nuclei independent chemical shift,NICS)方法。
NICS的定义是某个被置于待考察环中心的原子所感受到的磁屏蔽数值的相反数。由于其定义明确、考察方便,NICS成为当前研究芳香性的最重要和最标准的方法之一。由于没有任何化合物能有原子出现在环的正中心,NICS必须通过计算得到。具体过程为:在感兴趣的位置放置虚拟原子,进行核磁共振计算,从而得到该虚拟原子处的磁屏蔽张量和各向同性磁屏蔽数值。
对于芳香环,NICS表现为负值,并且一定程度上绝对值的大小与芳香性强弱呈现相关(但也不完全,特别是当几个芳环的富电子程度相差悬殊时,NICS容易受到环上电子密度因素的干扰)。而对于非方向性和反芳香性体系,NICS分别为接近0的数值和正值。NICS规定了作为探针的原子要放在待考察的环的中心,而至于沿着垂直于环平面的方向的位置,则可以视情况选择。人们一般取 NICS(1),即使得探针原子距离环平面1Å。在M06-2X/ def2-TZVP水平下,苯和环丁二烯的NICS(1)分别为一10.3 ppm和20.3 ppm,芳香性和反 芳香性的分野可见一斑。
通过改变探针原子的位置,可以通过NICS依次考察不同环的芳香性。以苯并环丁二烯为例(图4.13),在M06-2X/def2-TZVP水平下,苯和环丁二烯处的 NICS(1)分别为—2.9ppm和14.3 ppm, 说明苯环的芳香性被削弱了,而环丁二烯处则呈现出反芳香性。图4.13中作为探针的虚拟原子,位于两个环的正中心。
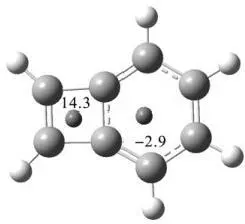
图4.13 苯并环丁二烯两个环处的NICS
除NICS外,Geuenich等提出的AICD(anisotropy of the induced current density)也是较为流行的通过磁学性质研究芳香性的方法。芳香性与反芳香性分子在外加磁场下分别产生抗磁环流和顺磁环流,AICD定义了一组反映感应电流方向和大小的函数。通常AICD的结果用等值面加箭头的方向展示,等值面表示了磁感应电流的大小,等值面越大的位置感应电流密度越大,反映了电子有较强的离域性;而箭头标识了磁感应电流的方向。AICD等值面需要基于Gaussian的核磁共振计算结果,结合单独的AICD程序处理得到。仍以苯并环丁二烯为例,在M06-2X/def2-TZVP水平下得到的AICD等值面如图4.14所示 。
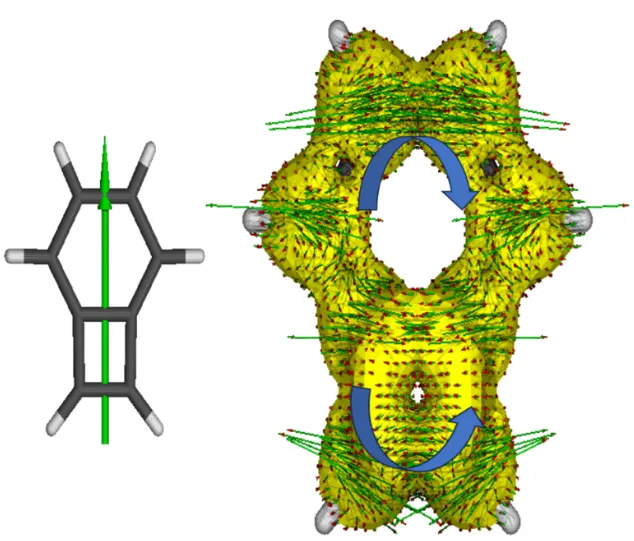
图4.14 苯并环丁二烯的 AICD 等值面
其中外加磁场的方向垂直于环平面向上。抗磁环流的方向遵循左手定则,让磁场方向通过左手大拇指,四指的方向即表示抗磁环流的方向。可以明显发现,苯和环丁二烯处的磁环流方向不同,前者遵循左手定则,可知苯和环丁二烯分别为芳香性和反芳香性的。
除了上述介绍的几种经典的用于研究芳香性的手段外,由于芳香性本质上来源于电子的离域,也可以借助键级、ELF等方法来辅助研究。利用键级进行研究,本质上与借助几何结构进行判断异曲同工;而如想通过ELF来研究,则最主要是观察沿着环是否有连续的ELF分布,如有,则意味着电子倾向于聚集到这个区域,并且在该区域内呈现出较大的离域性,而不容易出现在该区域之外,这与芳香性所对应的电子离域相吻合。此外,还有同样基于磁学效应的ICSS等多种办法。由于芳香性多维度、多角度、多内涵的特性,各种分析手段可以综合运用,从而获得关于体系电子结构的尽可能丰富的信息。

多位专家力荐 超全实战指南


↑扫码了解更多书籍及唯理计算信息








