









 您已经拒绝加入团体
您已经拒绝加入团体

 2025-07-02
2025-07-02
 8182
8182
 0
0
【摘要】 本文节选自唯理计算工程师团队所著《模拟计算指南》,深度解析原子电荷的划分原理与实战应用。涵盖Mulliken、Hirshfeld、NPA、CM5、AIM、RESP等电荷定义的特点及局限性(如Mulliken电荷的基组依赖性),并通过铵根离子(NH₄⁺)、氟化钠(NaF)等分子案例对比电荷分布。

关于本书
《模拟计算指南》是唯理计算工程师团队沉淀7年实战经验、历时一年打造,是一本计算化学快速入门指南、材料模拟计算领域的实用宝典。
“书中详细介绍了从理论计算化学的基本原理到目前国际前沿应用体系的计算模拟思路和方法,有利于读者从多维度理解如何采用理论计算方法来解决复杂科学问题,并帮助初学者从中找到适合自己科研的理论支持和计算解决方案。”
——教育部长江学者、杰青、复旦大学教授
刘智攀
“本书以其实用性和易学性为特色,无论是计算物质科学的初学者还是资深研究者,都能从中获得独特的视角和丰富的知识资源,使其成为该领域内一本极具价值的入门及参考书籍。”
——教育部长江学者特聘教授、华南师范大学教授
赵纪军

扫码了解更多书籍及唯理计算信息
01文章介绍
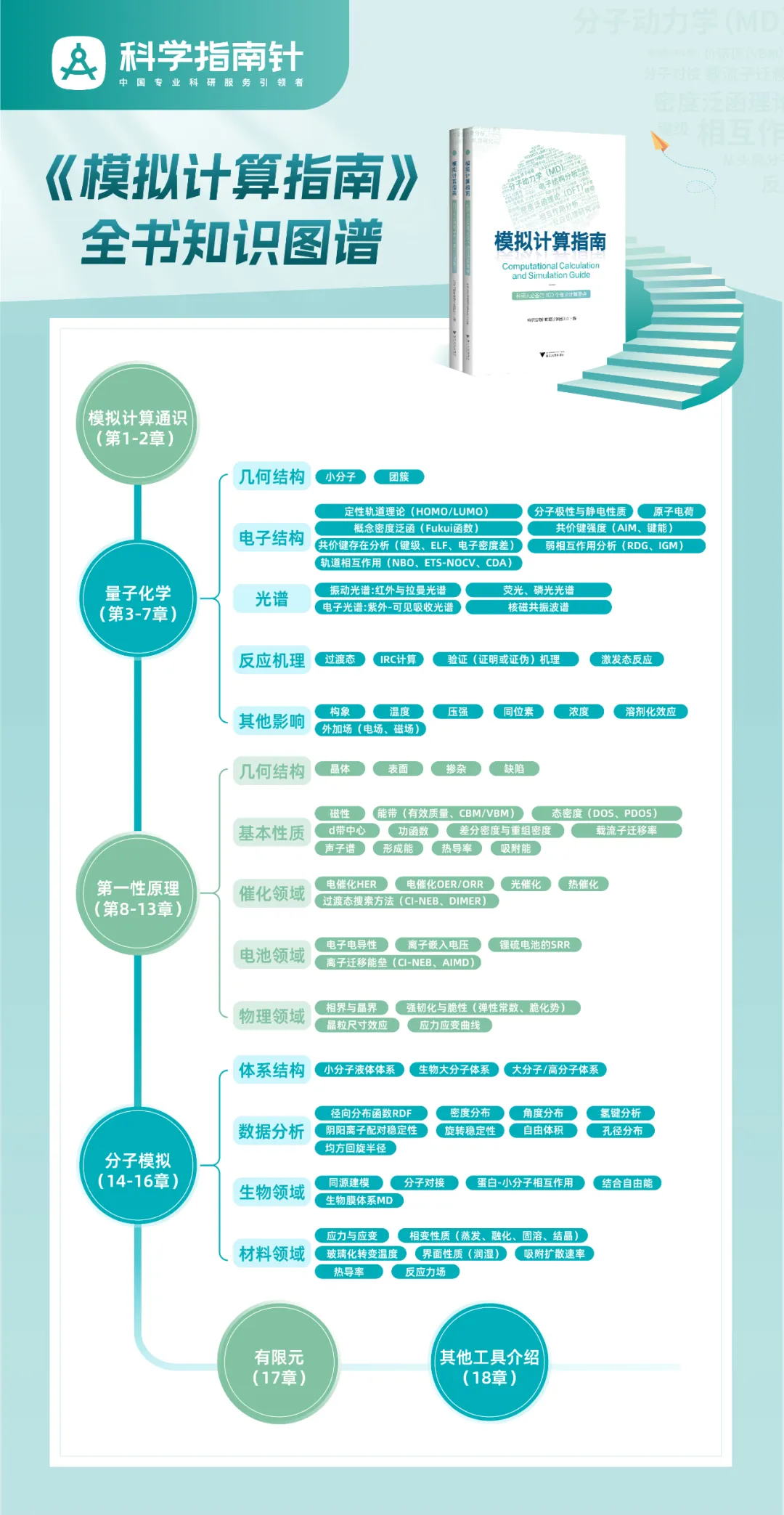
今天我们介绍下《模拟计算指南》的3.6.2原子电荷。
原子电荷的概念可理解为将分布于整个分子中的所有电子进行分派,划定出某个原子周围分布有多少电子,进而与核电荷数结合而得到它所分配得到的电荷。其中最关键的是对空间进行划分,给每个原子分配特定的区域,该区域内的电子即认为归属于该原子。依赖于划分方式,原子电荷有多种不同的定义,如Mulliken、Hirshfeld、NPA、CM5、AIM、RESP等。这些划分方式所基于的思想各不相同,人们在长期实践中,对其特点和实际表现也有了丰富的认识。以下简介几种常见的原子电荷:
(1)Mulliken电荷。Mulliken电荷的定义最为简单,在大多数量子化学程序中均有内置,随着SCF收敛就会直接输出。然而与此同时,Mulliken电荷的划分过于简单、生硬,导致其物理意义和实际表现均不理想,并且随着计算水平而变化很大,缺少实用价值。特别是当基组中带有弥散函数时,Mulliken电荷完全没有意义。
(2)Hirshfeld电荷和CM5电荷。Hirshfeld电荷在Gaussian中可以通过写入pop=hirshfeld关键字来输出,也可以基于fchk文件使用Multiwfn程序后处理得到。其物理意义与实际表现均很好,非常适合用于讨论电子结构和反应性。CM5电荷是Hirshfeld电荷的改进,数值普遍比Hirshfeld电荷更大,便于讨论和比较,同时对偶极矩的重现性也较好。Hirshfeld电荷和CM5电荷在定性上通常一致,都是广泛采用的原子电荷定义。
(3)NPA电荷。NPA电荷常被误写作NBO电荷,这是由于它是基于NBO方法得到的。NBO是一种轨道定域化方法,将在本书后续章节中介绍。对于普通有机分子,NPA电荷有很好的表现,但对于电子结构复杂、偏离经典路易斯结构的物质以及过渡金属配合物等适用性有限。为了得到NPA电荷,需要使用NBO程序。该程序在Gaussian中有内置,需要书写相应关键字。
(4)AIM电荷。AIM电荷又称作Bader电荷,是基于电子密度的零通量面对空间进行划分,从原理上十分严格,但实际表现却很不理想,时常出现与通常的化学认识以及实际反应性截然不同的情况,因而在量子化学计算中几乎不会使用。在第一性原理计算中,受到程序功能限制,AIM电荷比较流行。
(5)RESP电荷。RESP电荷是专门为了重现分子表面静电势等性质而设计的,主要用于构建分子动力学模拟所需的力场,很少用于讨论分子的反应性。
02实战示例
以下通过几个例子来进一步介绍原子电荷。
1. 铵根离子(NH₄+)
在铵根离子的路易斯结构式中,N上带有一个形式正电荷。但显然,由于N的电负性比H高得多,实际分子中正电荷必然集中在H上。在M06-2X/TZVP水平下,铵根离子的几种不同定义的原子电荷如表3.3所示。
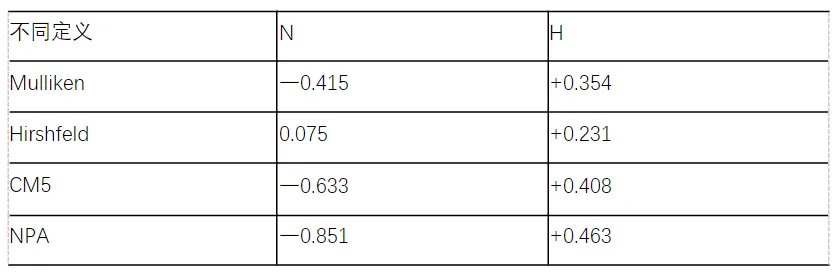
表3.3铵根离子在不同定义下的原子电荷
可以充分看到,所有原子电荷定义下,正电荷都集中在H上;除了Hirshfeld电荷外,各种原子电荷定义下,N都带有负电。这个例子充分说明了形式电荷与实际电荷是完全不同的事物。此外,虽然N和H的氧化态分别是-3和+1,但其实际电荷远没有这么夸张,都在零点几的数量级。
2. 氟化钠分子(NaF)
通常认为氟化钠是典型的离子化合物。是不是一个Na原子和一个F原子构成的双原子分子中Na和F的原子电荷就是+1和一1呢?这个问题的答案可以通过观察表3.4中的原子电荷得到。
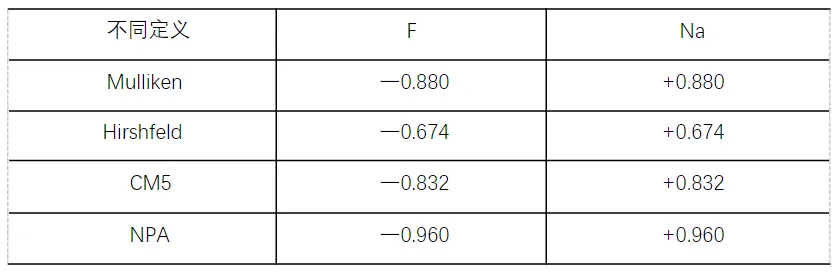
表3.4 NaF分子在不同定义下的原子电荷
可以发现,即使是NaF这种高度离子化的化合物,大部分定义下的原子电荷也都达不到形式上的氧化态(除了NPA电荷的数值比较大,这是由于它在原理上就更侧重于贴合路易斯结构式的定性描述)。这意味着Na—F之间的成键仍然有不可忽视的共价成分。
虽然原子电荷的自身数值不可能与氧化态相同、大多数情况下甚至不可能相近,但原子电荷的变化可以对氧化态的变化起到提示作用。同时,通过对分子中各片段的原子电荷进行求和,可以知道片段之间的电荷转移情况。如对于图3.23中TS5b和TS51两个过渡态,对N-N-NPincer配体上的CM5电荷进行求和,发现其数值分别为0.3547和0.3757,从而知道在TS51中配体向Co转移了更多的电子。因此我们就能预测,当在配体上引入吸电子取代基时,将有利于经由TS5b的过程,反之亦然。
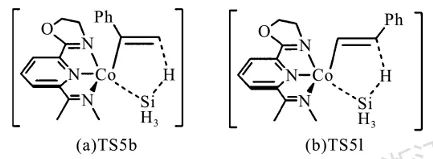
图3.23 两个过渡态的结构式
原子电荷对于判断化学反应选择性也有一定意义。一个经典的例子是芳香亲电取代反应,人们很早就发现其选择性与原子电荷存在关联。以苯胺和苯甲腈为例(图3.24),可见苯胺的2,4位碳原子具有较负的原子电荷,其中2位更负;而苯甲腈的3位碳原子电荷最负,4位次之。因此,如果亲电取代反应的选择性是静电主导的并且决速步为过渡态靠前的亲电试剂对芳环的加成,则可知苯胺的反应将主要发生在2位,而苯甲腈则主要发生在3位。
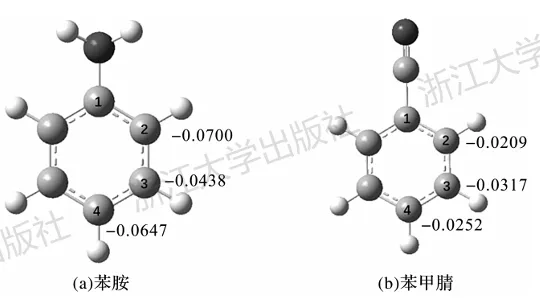
图3.24 苯胺和苯甲腈中部分原子的 Hirshfeld电荷
必须注意的是,上述推理的前提条件,即决速步为加成反应、过渡态靠前、静电主导,缺一不可!就上述亲电取代而言,过渡态靠前保证了底物的原子电荷等性质能够充分体现在决速步的过渡态中,这是使用底物性质讨论反应性的前提条件。为了满足这一条件,亲电试剂必须有足够高的亲电性,因此我们可以预测上述推理比较适用于硝化反应,而不适用于稳定碳正离子发生的傅克烷基化反应。静电主导的前提条件要求亲电试剂是硬酸,因此我们可以知道上述推理不适用于金属化反应。对于这类软酸作为亲电试剂的场合,轨道作用将成为决定性的因素。原子电荷还可以进一步告诉我们,亲电试剂越硬,则与苯胺反应时发生2位取代相比于4位取代优势越大;而对于苯甲腈的反应,则随着亲电试剂变硬,4位取代产物与2位取代产物的比例将逐渐增加。
因此,想要正确理解计算结果,必须对反应机理有深刻的思考和认识。对于每个化合物,其原子电荷等各种性质都是固定的,但能发生的反应却千差万别,随着条件不同,可以得到各种产物。如果不注意前提条件,想当然地用原子电荷或其他任何分子性质来机械化地预测反应产物,势必会得到错误的结果。

多位专家力荐 超全实战指南


扫码了解更多书籍及唯理计算信息








