









 您已经拒绝加入团体
您已经拒绝加入团体

 2025-07-08
2025-07-08
 8419
8419
 0
0
【摘要】 本节深入解析弱相互作用的类型(如氢键、π-堆叠、色散作用),强调色散作用的量子本质及其在分子稳定中的关键角色。介绍DFT方法(如DFT-D3)和可视化工具(RDG、IGM),通过六苯乙烷等实例展示色散作用对几何结构和能量的影响。内容涵盖理论原理、计算方法和实际应用,适合计算化学初学者和资深研究者参考。

关于本书
《模拟计算指南》是唯理计算工程师团队沉淀7年实战经验、历时一年打造,是一本计算化学快速入门指南、材料模拟计算领域的实用宝典。
“书中详细介绍了从理论计算化学的基本原理到目前国际前沿应用体系的计算模拟思路和方法,有利于读者从多维度理解如何采用理论计算方法来解决复杂科学问题,并帮助初学者从中找到适合自己科研的理论支持和计算解决方案。”
——教育部长江学者、杰青、复旦大学教授
刘智攀
“本书以其实用性和易学性为特色,无论是计算物质科学的初学者还是资深研究者,都能从中获得独特的视角和丰富的知识资源,使其成为该领域内一本极具价值的入门及参考书籍。”
——教育部长江学者特聘教授、华南师范大学教授
赵纪军

↑扫码了解更多书籍及唯理计算信息
01文章介绍
今天我们介绍下《模拟计算指南》的3.7色散作用及弱相互作用分析。
弱相互作用(weak interaction)是各类非键相互作用的统称,包含氢键、卤键、π-堆叠、π-阴离子(阳离子)相互作用、一般性的静电作用和色散作用等。色散作用(dispersion interaction)在其中占据重要的地位,因此本节首先对其进行介绍。
色散作用与通常所说的范德华作用有很大的重合度,但也不完全相同。习惯上所说的范德华作用泛指各种较弱的分子间作用,其本质包含了一部分静电、电荷转移、色散等多种相互作用类型,而色散在其中构成了绝大部分。传统上人们将范德华力中的“色散力”归属于瞬时偶极的相互作用,这一说法模糊了色散作用的本质,容易引发“色散作用属于静电作用”的误解。事实上,色散作用起源于量子力学中的电子相关作用,没有经典力学上的对应,这也给量子化学方法描述色散作用带来了很大的挑战。
高精度的post-HF方法(如耦合簇)可以很好地描述色散作用,是相关研究的金标准。在DFT方法中,多数早期泛函(如B3LYP、PBE0等)完全无法描述色散作用。为了解决这一问题,有两种路径。其一是在泛函形式上做文章,在设计泛函时就设法拟合色散作用主导的体系,以求使得泛函本身就能够描述色散作用。随着色散作用逐渐受到关注,大部分较为现代的泛函,如M06-2X、M11、MN15等在提出时考虑了色散作用,因此具备较好的描述色散作用的能力。另一条道路则是在现有泛函的基础上引入校正项。校正项的形式各有不同,目前最流行的是Grimme提出的DFT-D方法。
DFT-D方法在DFT计算得到的能量基础上加入一个经验拟合的能量项,以表现色散作用的能量贡献。随着方法的发展,目前已经出现了DFT-D2、DFT-D3和DFT-D4三个版本,每个版本都比前一个有所改善。当前DFT-D3由于在主流程序中支持广泛而最为流行,而更好的DFT-D4也在不断普及。DFT-D2和DFT-D3的校正项均只依赖于原子坐标,不会改变SCF迭代的过程,因此几乎不会造成耗时增加。DFT-D4在此基础上进一步考虑了原子不同带电状态下色散作用的差异。对于大部分主流泛函,DFT-D都拟合了相应参数,可以直接搭配使用。常见的诸如B3LYP-D3BJ等的写法,就是指在B3LYP的基础上使用了DFT-D3,具体来说是其中使用BJ阻尼的形式(另一种形式是零阻尼)。
除了对于B3LYP等完全无法描述色散作用的泛函可以使用DFT-D外,针对M06-2X等本身已经能够描述色散作用的泛函,DFT-D也有相应的参数,结合使用号称可以进一步改善其描述能力,但实际表现则因体系而异。目前来看,对于B3LYP、PBE0等泛函,推荐一律结合DFT-D使用;对于M06-2X等,则往往可以不用。此外,wB97xD泛函自身已经内置了DFT-D,无法再加。
色散作用对几何结构和能量的影响通常是不可忽略的。一方面,卤键、π-堆叠、π-阴离子相互作用等绝大部分都由色散作用贡献,如果忽略了色散作用,甚至将无法得到正确的几何构型;另一方面,当体系较大、包含长链烷基等时,基团间的色散作用也将对几何结构和能量带来影响。
为了直观了解色散作用的重要性,可以考察一系列色散稳定的分子。其中最知名的是取代的六苯乙烷。六苯乙烷母体高度不稳定,无法分离得到,通常人们认为这是由于六个苯基间巨大的立体排斥所致。然而在1986年Mislow却令人惊讶地分离到了六(3,5-二叔丁基苯基)乙烷[16]。十二个大体积的叔丁基的引入出人意料地使得这个分子被稳定化了,并且能够在熔点(214℃)附近保持稳定(图3.25)。Grimme等人在2011年阐述了这一分子得以存在的原因[¹7,通过详细的计算论证,阐明了正是大体积烷基之间的色散相互作用使得该物质得以稳定存在。通过DFT-D方法,可以很好地描述此类体系中的色散作用。
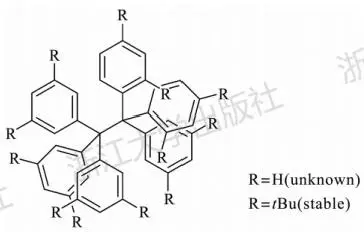
图3.25六苯乙烷的结构式
在合理考虑色散作用的基础上,可以使用量子化学计算的工具探讨各类弱相互作用的存在性、强度及本质。探讨弱相互作用的强度最重要的方法是设计相关的结合过程,并计算其能量变化。需要注意的是,结合过程不一定由单一的弱相互作用贡献,如可能既有氢键,也有π-堆叠、一般性的色散作用;可能有多重氢键,同时存在多种相互作用等,这些共存的相互作用的能量贡献是无法分离开的。
除了一个复合物中可能有多种弱相互作用外,一个弱相互作用中,也可能有多种不同本质的相互作用共同影响,这些影响因素包括色散作用、静电作用、Pauli交换作用、电荷转移等。在不同种类的弱相互作用以及同种但不同体系的弱相互作用中,起主导作用的因素各不相同。以氢键为例,多数情况下,经典强氢键主要由静电作用贡献,而随着氢键强度减弱,色散等其他因素的贡献逐渐增加。为了探讨弱相互作用的贡献因素,可以借助能量分解的手段。
为了验证弱相互作用的存在性并进行可视化,可以借助RDG(reduceddensitygradi-ent)[18]、IGM(independentgradientmodel)1⁹等方法。这些方法均以等值面填色图的方式呈现,旨在可视化地展示弱相互作用存在的区域,并通过颜色来对应弱相互作用的强度和本质。
RDG等值面可以基于量子化学计算得到的波函数文件使用Multiwfn程序处理得到。其中等值面呈现片状,绿色和蓝色分别表示较弱和较强的吸引作用,红色表示排斥作用。以一个基于π-堆叠而组织起来的三聚体为例(图3.26),可见芳环间存在大片绿色的等值面区域,结合几何构型中芳环平行排列的事实,可以清晰地知道π-堆叠作用的存在性。此外,在一些O—H附近存在蓝色的等值面,结合几何构型符合氢键的特点,可知存在较强的氢键作用。在苯环中心等处存在许多红色的等值面,提示排斥作用,这是由于各片段内部受到分子骨架限制有许多原子靠近,与所感兴趣的问题无关。
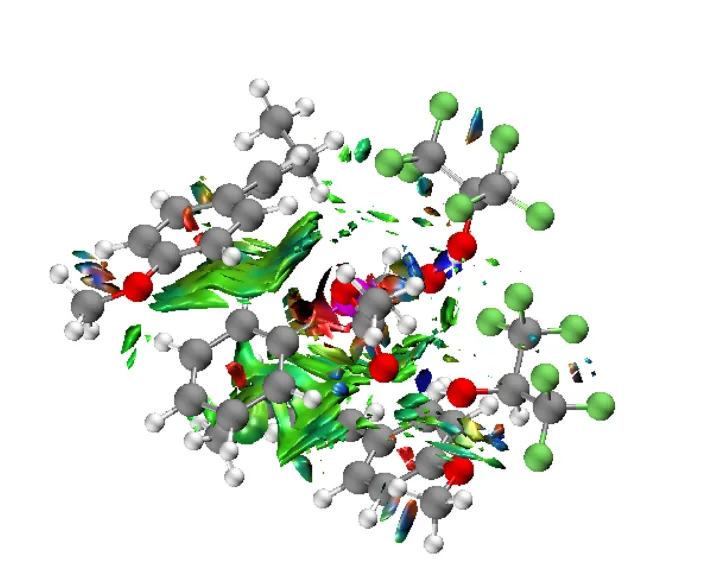
图3.26一个复杂三聚体分子的RDG等值面填色图
RDG可以体现出弱相互作用的空间分布,但有着等值面过于琐碎的缺点,并且不单单体现了片段间的作用,许多不感兴趣的片段内作用也被展示了出来,上文提到的环中心排斥作用就是一例。IGM分析与RDG含义非常类似,但在得到IGM等值面的过程中,需要指定如何划分片段,随后可以只展示给定片段间的相互作用,使得最终图像更加清晰。针对上文中的三聚体,恰当选定片段后,通过IGM即可清晰地展示出特定两个苯环之间的π-堆叠作用(图3.27)。
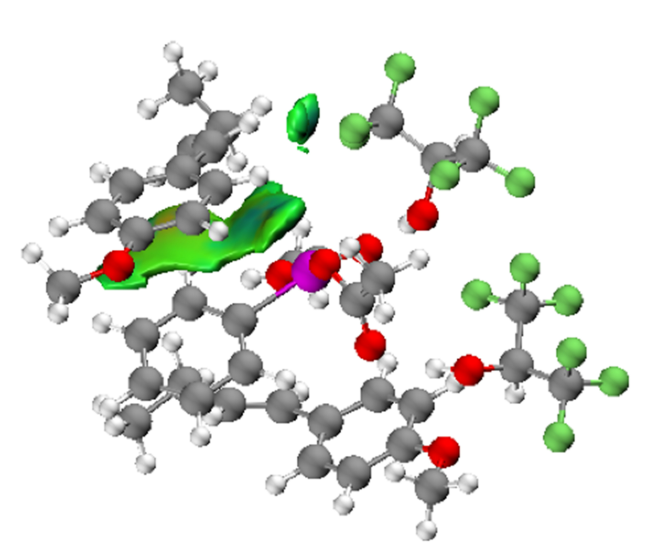
图3.27一个复杂三聚体分子中特定片段间的IGM等值面填色图
需要注意的是,RDG、IGM等可视化手段,终究是用于验证弱相互作用存在性的辅助手段,想要判断某种弱相互作用是否存在,最关键的仍然是对几何结构和化合物性质的认识。观察几何结构是否符合弱相互作用的要求,是判断弱相互作用存在性的第一步和最重要一步,只有在几何结构符合相关相互作用的定义的基础上,后续讨论才有价值,切莫犯因为不了解弱相互作用的化学本质和几何要求而将一种弱相互作用当作另一种的错误。

多位专家力荐 超全实战指南


↑扫码了解更多书籍及唯理计算信息








