









 您已经拒绝加入团体
您已经拒绝加入团体

 2026-07-14
2026-07-14
 25
25
 0
0
【摘要】 广州大学秦冬冬、牛利团队相关研究发表于 ACS Energy Letters,首次报道结晶型 Ru(II) 配合物量子点用于光催化析氢,解决脂溶性钌配合物水分散性差、团聚严重和电荷扩散受限问题。科学指南针唯理计算助力 DFT 理论计算,包括 HOMO/LUMO 前线轨道分布、激发态能级和自旋轨道耦合 SOC 分析,揭示 ³MLCT 激发态形成、界面电荷转移和高效析氢机制。


期刊:ACS Energy Letters
IF:17.5
▎唯理计算 · 本项目技术支持
密度泛函理论(DFT)计算,包括前线分子轨道(HOMO/LUMO)分布、激发态能级、自旋轨道耦合。
▲共同第一作者:田欣,孙春霖,佟连鹏
共同通讯作者:秦冬冬,牛利
通讯单位:广州大学
论文DOI:10.1021/acsenergylett.5c04176
这项发表于 ACS Energy Letters 的研究报道了结晶型钌(II)配合物量子点用于光催化析氢的新策略。研究通过表面活性剂辅助乳化,将三类脂溶性 Ru(II) 联吡啶配合物转化为粒径 2.8–4.8 nm、水分散性良好的结晶量子点,解决了传统钌配合物在水相中易团聚、结晶性差和电荷扩散受限的问题。其中 RuQT-3-QD 在 AM 1.5G 模拟太阳光下实现 15.62 mmol·g⁻¹·h⁻¹ 的析氢速率。科学指南针唯理计算助力完成该研究中的 DFT 理论计算,包括 HOMO/LUMO 前线轨道分布、激发态能级和自旋轨道耦合 SOC 分析,为揭示 ³MLCT 激发态形成、界面电荷转移和高效光催化析氢机制提供了理论支持。
全文速览
本研究合成了三类光物理性能与电子结构可调的脂溶性钌(II)配合物,并将其转化为粒径2.8-4.8 nm的高水分散性结晶量子点,解决了钌(II)配合物普遍较差的水溶性造成水相团聚尺寸与电荷扩散距离不匹配的问题。在AM 1.5G模拟太阳光(λ>420 nm)条件下,系统测试该量子点的光催化析氢性能,讨论了三重态金属-配体电荷转移(MLCT)激发态的界面电荷交换动力学行为。该结晶量子点的最优析氢速率高达15.62 mmol·g⁻¹·h⁻¹,远超仅以钌(II)配合物为单一活性组分的已报道光催化剂,这也是首例结晶型钌(II)配合物量子点用于光催化析氢的研究报道。
背景介绍
以太阳光为能源、水为原料的光催化析氢反应,是极具应用前景的氢能制备技术之一。半导体催化剂作为反应核心位点与能量转化单元,其光捕获能力、光生电荷浓度与能级、体相载流子扩散路径与速率,以及多电子界面反应动力学,直接决定整体催化效率。钌(II)联吡啶配合物凭借p型导电特性,以及金属-配体电荷转移(MLCT)主导激发态的热力学适配能级,成为光催化析氢的优异候选材料;钌的重原子效应可强化自旋-轨道耦合,大幅提升系间窜越(ISC)效率,进而形成高浓度、长寿命的三重态激发态,为高效析氢奠定核心基础。更为关键的是,联吡啶衍生物配体的π反键轨道能级低于钌(II)中心的eg*轨道,从根源上抑制了破坏配合物稳定性的金属中心(MC)激发态形成。
钌(II)联吡啶配合物作为光敏剂在染料敏化太阳能电池中应用成熟,但在光催化析氢领域的应用仍十分有限。目前相关研究多将其用作分子催化剂,或与宽禁带半导体复合用于光解水,也有研究将其嵌入共价有机框架(COF)、金属有机框架(MOF)调控析氢电荷转移路径,双核钌(II)配合物、特殊配位结构钌(II)配合物也分别在二氧化碳还原、水氧化反应中得到探索。结合钌(II)配合物本征特性与光催化水解基础原理,制约其高效析氢的核心瓶颈有两点:一是配合物亲脂性较强,在水相中分散不均、分子团聚严重,引发激发态淬灭与电荷过度复合;二是材料结晶性差、分子堆积无序,阻碍电荷扩散与表面催化反应。因此,开发兼具水溶性、小尺寸、高结晶性,且保留高最低未占分子轨道(LUMO)能级与长寿命激发态本征优势的钌(II)配合物聚集体,成为该领域的核心研究需求。
本文亮点
本研究合成了三种光物理性质可调的脂溶性钌(II)联吡啶配合物(RuQT-1/2/3),通过表面活性剂辅助乳化策略,成功将其转化为粒径2.8-4.8 nm的水分散结晶型量子点,并将其应用于光催化析氢领域。本工作的创新点展现在:第一,这是结晶型钌(II)配合物基量子点的首次报道,该结构形式同步解决了传统材料的水分散性与结晶性双重难题,所得量子点尺寸均小于5 nm,结晶性能优异且表面亲水性良好(水接触角6.6°-19.4°),可有效助力光吸收、电荷分离与质子转移过程;第二,在AM 1.5G模拟太阳光(100 mW/cm²,λ>420 nm)照射下,LUMO在三配体中均匀离域、与水相氧化还原物种电荷交换性能最优的RuQT-3量子点,析氢速率可达15.6 mmol·g⁻¹·h⁻¹,该数值是经典水溶性Ru(bpy)₃Cl₂催化剂的120倍,也是纯钌(II)配合物作为单一活性组分体系中已报道的最高值;第三,本研究提出的攻克脂溶性分子水分散性与结晶性瓶颈的光催化改性策略具备普适性,除钌(II)配合物外,还可拓展应用于其他疏水性离子型分子。
图文解析
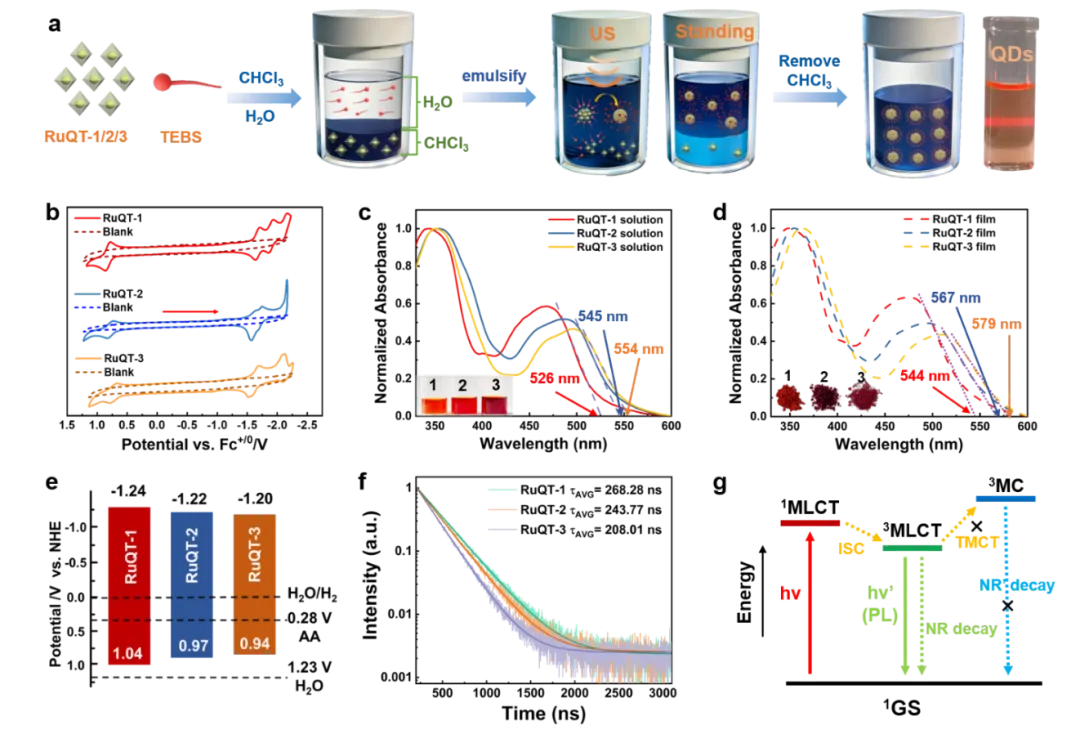
Figure 1.(a) Schematic illustration of the synthesis process of water-soluble quantum dots of three complexes. (b) Cyclic voltammetry (CV) curves of the complexes in 1.0 mM acetonitrile (CH₃CN) solution. (c-d) UV-vis absorption spectra of the complexes in 1×10-5 mol·L-1CH₃CN solution (c) and solid film (d) state. (e) Experimentally determined redox potentials of the complexes. (f) Room-temperature phosphorescence lifetime of the complexes in 1×10-5 mol·L-1CH₃CN solution. (g) Jablonski energy dagram accompanied by the orbtial of different states.
三种配合物均呈现两处特征吸收峰:RuQT-1的最大吸收峰位于341 nm和467 nm,RuQT-2为356 nm和488 nm,RuQT-3为351 nm和498 nm。其中短波峰归属于配体内部电荷转移(¹ILCT)跃迁,长波峰则为特征性的单重态金属-配体电荷转移(¹MLCT)跃迁。三种配合物的薄膜吸收谱图走势与乙腈溶液中相近,仅¹MLCT吸收峰出现8-11 nm的红移;且随着配合物中dthbpy配体数量增加,薄膜¹MLCT吸收截止波长发生红移。结合循环伏安法测得的最高占据分子轨道(HOMO)能级,以及对应光学带隙,构建了配合物的能级分布图,其足够负的还原电位,为光催化析氢提供了强劲的热力学驱动力。相较于溶液态,固态薄膜的发射峰红移27-39 nm。吸收与发射光谱的红移现象说明固态下存在分子间相互作用,但三种配合物均未出现平面有机分子常见的聚集诱导猝灭(ACQ)现象。
三种配合物在溶液与固态下具有较长的发射寿命。该寿命数值远高于已报道的有机小分子及聚合物光催化剂(通常仅为数纳秒),甚至优于含钌配合物的共价有机框架材料。较长的激发态寿命印证了三重态跃迁的存在,可让更多载流子参与光催化反应,而非通过非辐射过程失活。三种配合物均具备室温磷光(RTP)特性,磷光峰分别位于652 nm、680 nm、700 nm,寿命为208.01-268.28 ns。显著的室温磷光行为证明,配合物可通过高效系间窜越形成长寿命三重态金属-配体电荷转移(³MLCT)激发态,这是实现高效光催化的核心条件。
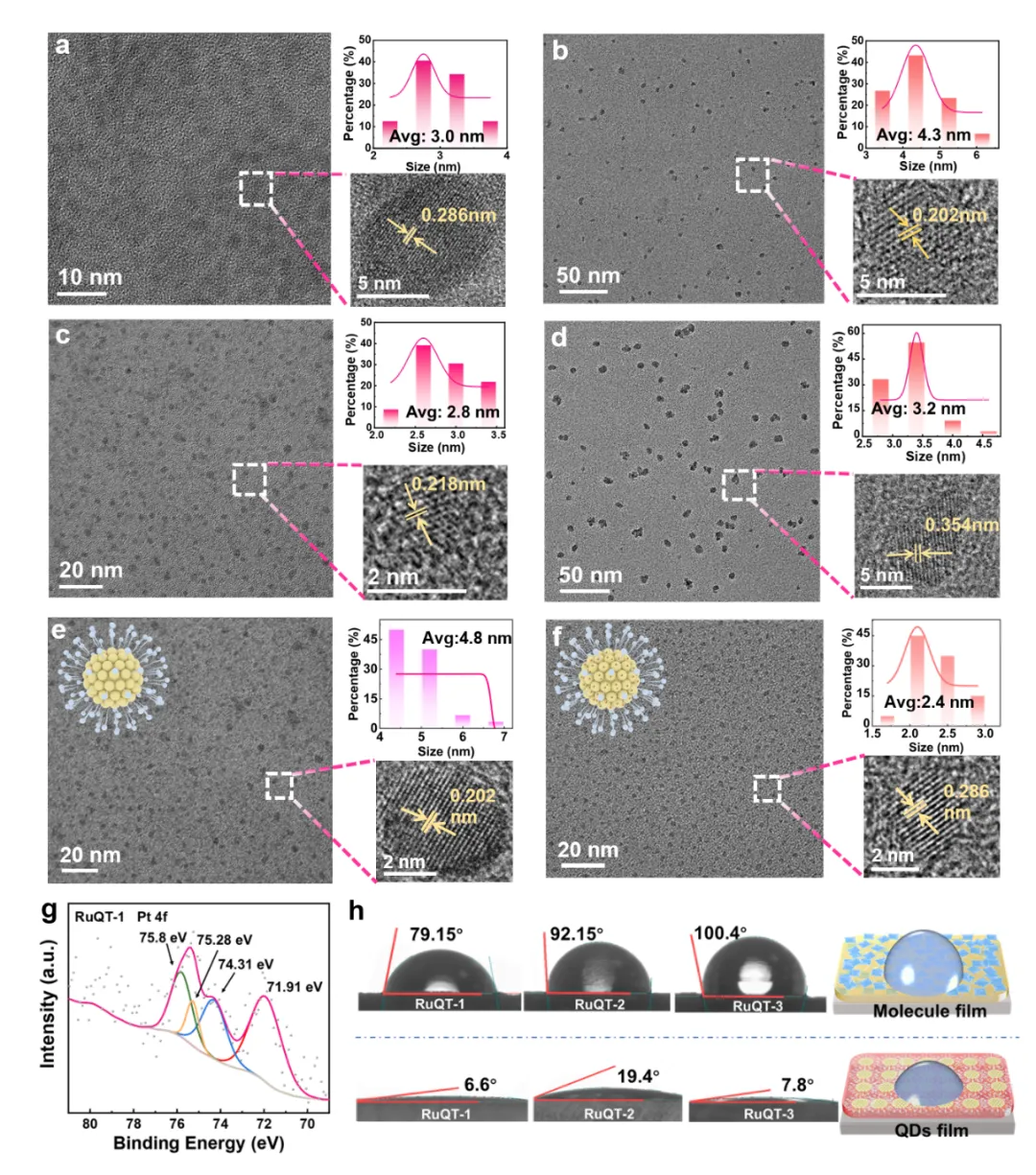
Figure 2. (a-f) Transmission electron microscopy (TEM) images of complex QDs: (a, c and e) RuQT-1/2/3-QDs without Pt modification, respectively and (b, d and f) with Pt modification. (g) X-ray photoelectron spectroscopy (XPS) Pt 4f spectra of Pt-modified RuQT-3-QDs. (h) Water contact angles (WCAs) of the molecular complexes and their corresponding QD films.
透射电子显微镜(TEM)显示三种配合物制备的量子点尺寸分布均一,粒径均小于5 nm,平均粒径分别为RuQT-1-QD 3.0 nm、RuQT-2-QD 2.8 nm、RuQT-3-QD 4.8 nm。在量子点表面负载铂后,其形貌与粒径未发生明显变化,粒径维持在2.4-4.3 nm。材料尺寸、载流子扩散距离与光吸收能力的最优平衡,是提升光催化效率的关键,钌(II)配合物具备高摩尔消光系数,可在小尺寸穿透深度内充分吸收入射光子;同时量子点的小尺寸特性大幅缩短光生载流子从体相到催化剂表面的传输距离,最大化提升表面载流子浓度,进而强化光催化析氢性能。高分辨透射电子显微镜(HR-TEM)图像可观测到清晰的晶格条纹,晶面间距为0.202-0.354 nm,证明量子点纳米域内分子有序堆积,规整的微区结构可有效加速电荷扩散。TEM图像中未观测到明显铂颗粒,说明铂负载量较低,且形成了超小尺寸铂物种。X射线光电子能谱(XPS)证实量子点表面的铂以金属态铂(Pt⁰)和氧化态铂(Pt²⁺)混合价态存在,该混合价态可发挥催化协同作用:金属态铂可加速电荷快速传输,Pt²⁺则可作为催化循环中的氧化还原中间体。水接触角测试结果显示三种原始配合物的水接触角为79.15°-100.4°,符合其疏水性本质。与之形成对比,三种量子点薄膜的水接触角大幅降至6.6°-19.4°,证实疏水性配合物已成功转化为亲水性胶体,优异的亲水性可大幅加速光生电荷与质子的转移速率,进一步提升光催化效率。
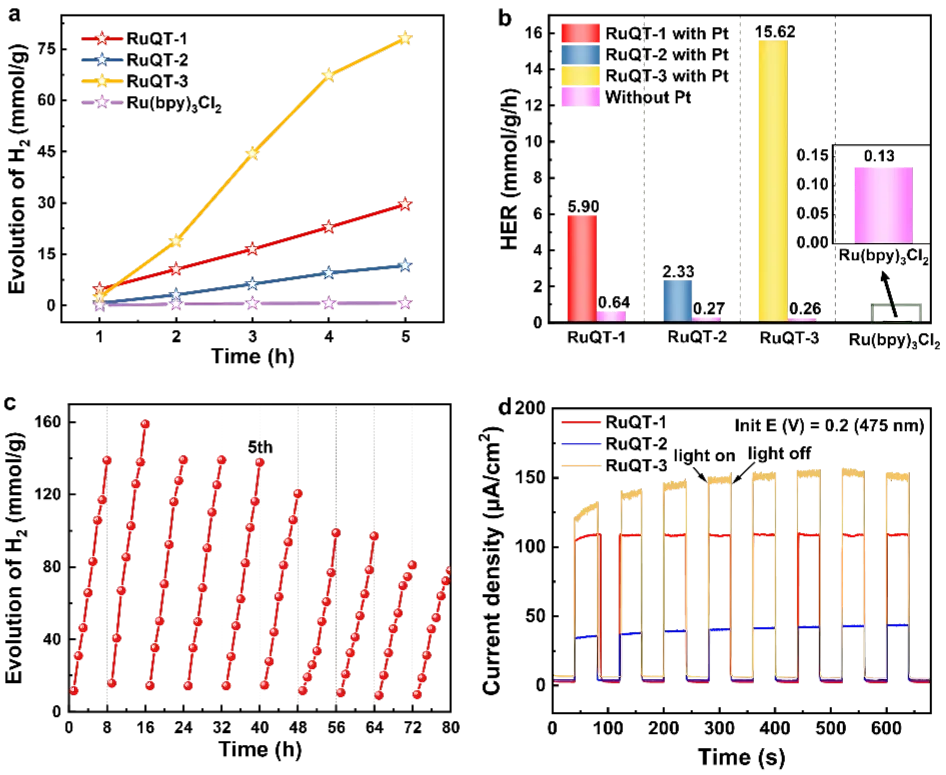
Figure 3. (a) Hydrogen production amount of QDs as a function of reaction time; (b) hydrogen evolution rates of the QDs; (c) long-term stability of the RuQT-3-QDs over 80 h photocatalytic reaction; (d) photocurrenti-t curve under chopped light (475 nm) at a bias potential of 0.2 V, measured in an electrolyte containing 0.1 M Na2SO4 and 0.1 M ascorbic acid (AA).
将三种配合物直接作为粉末态催化剂用于光催化析氢反应时,未检测到析氢信号;与之相对,其对应的量子点则展现出显著的光催化活性,远高于经典水溶性Ru(bpy)₃Cl₂。铂负载量为5.2 wt%的优化条件下,三种量子点均能在5小时内保持稳定且近乎线性的析氢动力学,析氢速率显著提升,分别为RuQT-1-QD 5.9 mmol·g⁻¹·h⁻¹、RuQT-2-QD 2.33 mmol·g⁻¹·h⁻¹、RuQT-3-QD 15.62 mmol·g⁻¹·h⁻¹。当将共催化剂换为金时,RuQT-1/2/3量子点的析氢速率分别为5.83、8.89、10.84 mmol·g⁻¹·h⁻¹,虽略低于铂助催化体系,但证实金同样可作为高效助催化剂,显著提升量子点的光催化析氢效率。
低自旋t2g6闭壳层构型带来的高配体场稳定化能(LFSE),赋予了配合物优异的长期稳定性。RuQT-3-QD在40小时反应(5次循环,单次8小时)后,催化活性几乎完全保留,超过40小时后析氢效率开始缓慢下降。反应80小时后补充空穴牺牲剂抗坏血酸(AA),无法恢复RuQT-3-QD的催化性能。采用标准三电极光电化学(PEC)电池,将配合物分子负载于垂直定向的金红石型TiO₂纳米棒上制备光阳极,三种配合物在可见光(λ>420 nm)及475 nm单色光照射下均产生明显光电流,证实了配合物的可见光响应特性及光电转换可行性。电化学阻抗谱(EIS)测试显示强亲脂性基团不利于配合物分子与电解液间的电荷转移。
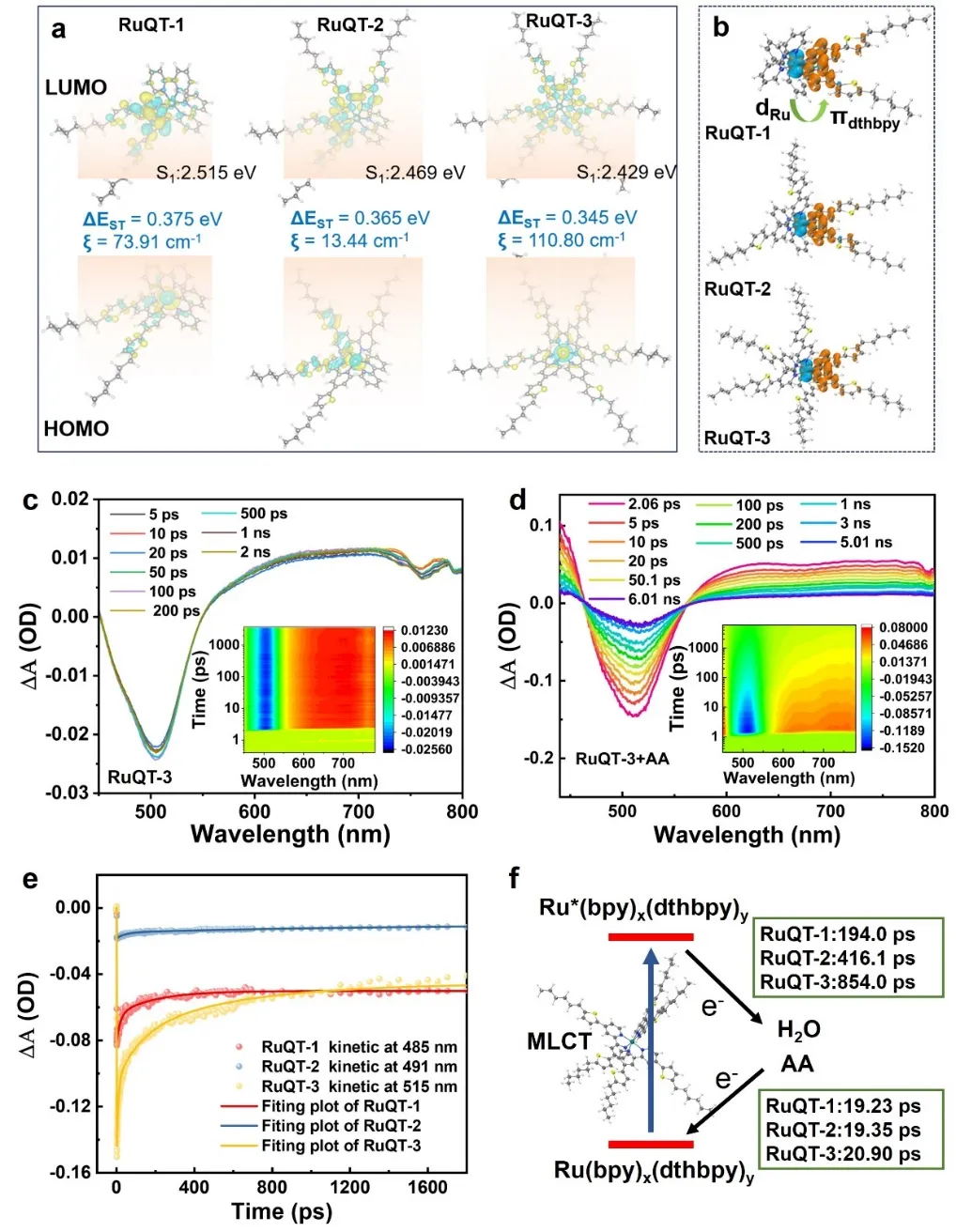
Figure 4. (a) Highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) distributions of the complexes in excited singlet state, with the isovalue=0.03; ΔEST represents the energy gap between the lowest singlet excited state (S₁) and the lowest triplet excited state (T₁), and ξ denotes the corresponding spin-orbit coupling (SOC) constant. (b) Hole (blue) and electron (red) distributions in the excited triplet state.(c, d) Femtosecond visible transient absorption spectra of the RuQT-3-QDsin water(1 mg in 30 mL water) in absence (c) and presence (d) of 1.0 mM AA. (e) Kinetics of the 485-515 nm for complexes’ QDs + AA aqueous system. (f) Schematic representation of exciton decay and electron/energy transfer processes of the complexes’ QDs + AA system.
理论计算结果表明,三种配合物的单重激发态中,空穴均局域在最高占据分子轨道(HOMO),主要呈现钌d轨道特征;电子则局域在最低未占分子轨道(LUMO),且主要分布于dthbpy配体而非bpy配体上。三种配合物的单重-三重态(S1-Tn,n=1-10)能隙较小,且S1与Tn态间的自旋-轨道耦合(SOC)常数较大,为高效系间窜越(ISC)提供了条件。高效系间窜越可大幅提升第一三重激发态(T₁)布居数,这是实现高效光催化的关键。数据进一步证实,S1→Tn跃迁(尤其是S1→T1跃迁)主要源于同类型轨道主导的金属-配体电荷转移(MLCT)过程。三重激发态下的空穴-电子分布结果进一步验证了配合物三重态的MLCT特征,同时确定催化活性位点局域于³MLCT态的dthbpy配体上。
三种配合物的量子点水溶液,飞秒瞬态吸收光谱(fs-TAS)特征相近:均出现485-515 nm处的宽频基态漂白峰(GSB)、540–600 nm处归属于激子吸收的弱光诱导吸收峰(PIA),且2 ns内光谱衰减可忽略。加入空穴牺牲剂抗坏血酸(AA)后,三种配合物的基态漂白与光诱导吸收信号均快速衰减,证明激发态配合物可发生超快激子空穴转移(<20.9 ps),空穴快速注入牺牲剂的同时伴随电子快速回传,利于维持水解还原半反应的稳态进行。基态漂白恢复过程可反映整体电荷分离动力学,涵盖³MLCT激发态电子向水的转移(还原过程)与空穴向牺牲剂的注入(氧化过程)。配合物³MLCT激发态向水的电子转移速率,远快于其室温磷光衰减速率(>208 ns)与常规钌(II)联吡啶配合物的非辐射衰变速率。综合动力学过程下,2 ns时间窗口内三种量子点的基态漂白恢复率分别为RuQT-1 37.5%、RuQT-2 44.4%、RuQT-3 66.7%。
总结与展望
本研究用于制备结晶量子点的钌(II)配合物的结构较为简单,在攻克水分散性与结晶性核心瓶颈后,已展现出优异的光催化析氢性能。由此可预见,以光催化析氢基本原理为指导,借助高效溶液合成手段,在原子层面精准设计并调控钌(II)配合物的分子组成、电子结构与电荷转移路径,有望进一步大幅提升该类配合物的光催化析氢效率。
FAQ
1.Ru(II) 配合物为什么适合光催化析氢?
Ru(II) 联吡啶配合物具有良好的可见光吸收能力和金属-配体电荷转移 MLCT 激发态特征。钌元素的重原子效应能够增强自旋轨道耦合,促进单重态到三重态的系间窜越,形成长寿命 ³MLCT 激发态,从而为光生电荷参与析氢反应提供更充足的时间窗口。
2.为什么要将脂溶性 Ru(II) 配合物制备成结晶量子点?
传统脂溶性 Ru(II) 配合物在水相中容易团聚,导致激发态淬灭、电荷复合增加和质子转移受限。将其制备成小尺寸、高水分散性、结晶型量子点后,可以同时缩短光生载流子扩散距离、改善水相接触界面,并通过有序分子堆积提升电荷传输能力,从而显著提高光催化析氢效率。
3.科学指南针唯理计算在这项 ACS Energy Letters 研究中提供了哪些支持?
科学指南针唯理计算助力完成了该研究中的 DFT 理论计算,包括 Ru(II) 配合物的 HOMO/LUMO 前线分子轨道分布、激发态能级和自旋轨道耦合 SOC 分析。这些计算用于解释配合物的 MLCT 电荷转移特征、三重态激发态形成机制、活性位点分布以及 RuQT-3-QD 高效析氢性能的理论来源。
4.SOC 自旋轨道耦合计算说明了什么?
SOC 计算用于判断单重激发态与三重激发态之间发生系间窜越的可能性。该研究中,Ru(II) 配合物的 S₁ 与 Tₙ 态之间能隙较小,并具有较大的 SOC 常数,有利于高效 ISC 过程,进而形成长寿命 ³MLCT 激发态。这是量子点能够实现高效光催化析氢的重要理论依据。
5.为什么 RuQT-3-QD 的析氢性能最好?
RuQT-3 的 LUMO 在三配体中分布更均匀,有利于与水相氧化还原物种进行界面电荷交换。结合瞬态吸收结果,RuQT-3-QD 在 2 ns 时间窗口内表现出更高的基态漂白恢复率,说明其电荷分离和转移动力学更优。因此,RuQT-3-QD 实现了 15.62 mmol·g⁻¹·h⁻¹ 的高析氢速率。
6.这项研究对 Ru(II) 配合物光催化有什么意义?
这项研究首次报道了结晶型 Ru(II) 配合物量子点用于光催化析氢,解决了传统钌配合物水分散性差、聚集态无序和电荷扩散受限的问题。该策略为疏水性分子光催化剂的水相应用提供了新路径,也为通过分子结构设计、激发态调控和量子点化改性提升光催化析氢效率提供了参考。








