









 您已经拒绝加入团体
您已经拒绝加入团体

 2022-10-08
2022-10-08
 6376
6376
 0
0
【摘要】 将四氢呋喃(THF)转化为高附加值的化学品是一种非常有前景的方法,而四氢呋喃固有的化学惰性使得其直接、选择性C(sp3)−H键活化非常困难。

背景
将四氢呋喃(THF)转化为高附加值的化学品是一种非常有前景的方法,而四氢呋喃固有的化学惰性使得其直接、选择性C(sp3)−H键活化非常困难。
使用化学计量氧化剂和氢原子转移试剂活化THF的α-C-H键在原子经济性、可控性和安全性等方面都有较大的限制。
结果与讨论
中科院理化技术研究所吴骊珠院士等报道半导体量子点能够在氧化还原中性的温和条件下,利用可见光选择性活化THF的α-C-H键。
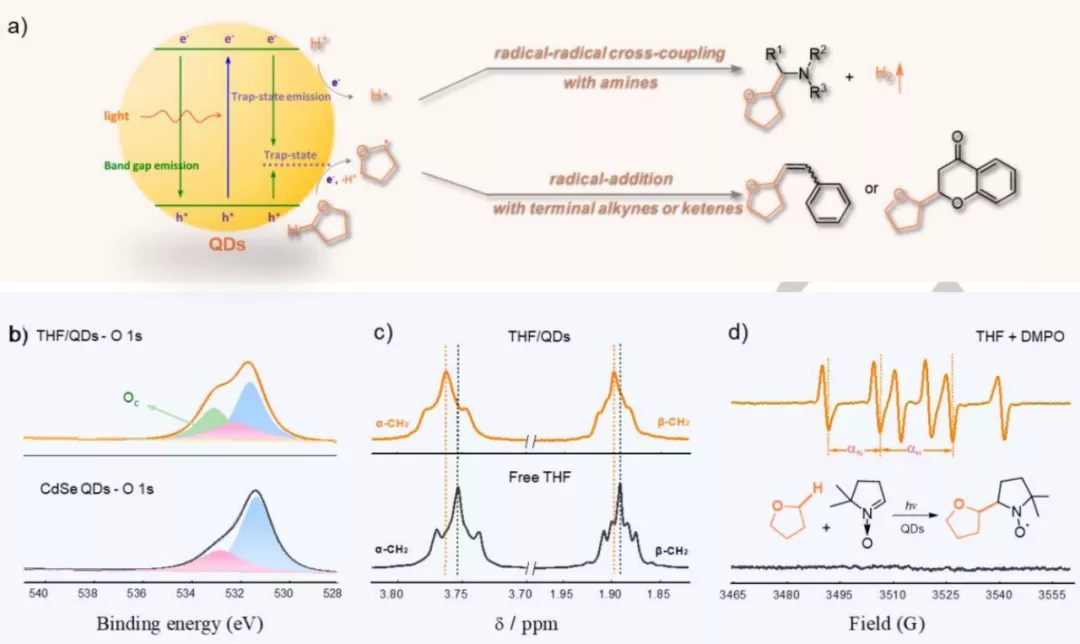
图1:(a)量子点表面THF的活化机理推测。(b) 游离THF和QDs/THF共轭结构的高分辨率XPS O1s光谱。(c)CDCl3中游离THF和THF/QDs共轭结构的1H核磁共振谱。(d)在室温和Ar氛围下,CH3CN/THF(v/v=6:1)、硒化镉量子点(2.9×10-5M)溶液的电子顺磁共振图谱。
如图1a所示,THF的氧原子和CdSe量子点的空位点结合,形成THF/QDs共轭结构,选择性地削弱THF的α-C-H键。
在可见光照射下,量子点表面的THF的α-C-H键会失去一个质子和电子生成烷氧基烷基自由基。
随后该自由基与氨基C(sp3)-H键产生的α-氨基自由基发生交叉偶联反应,或者分别与与烯烃或苯乙炔发生加成反应。
独特的THF/QDs共轭结构也通过XPS和核磁共振进行了表征,如图1b-c所示,THF/QDs共轭结构与游离THF的高分辨XPS O1s图谱相比,533.1 eV处的峰可以归为THF/QDs共轭结构内THF的化学吸附氧的峰。
与游离THF相比,THF/QDs共轭结构的1H NMR图谱化学位移向低场移动,证实了THF的α-C-H键的延长。
通过黑暗及光照条件下的电子顺磁共振(EPR)图谱(图1d)可以观察到,在黑暗条件下,QDs与THF共同存在并不能使得THF转化为烷氧基烷基自由基,只有在光照的驱动下,才能使得THF转化为自由基参与后续反应。
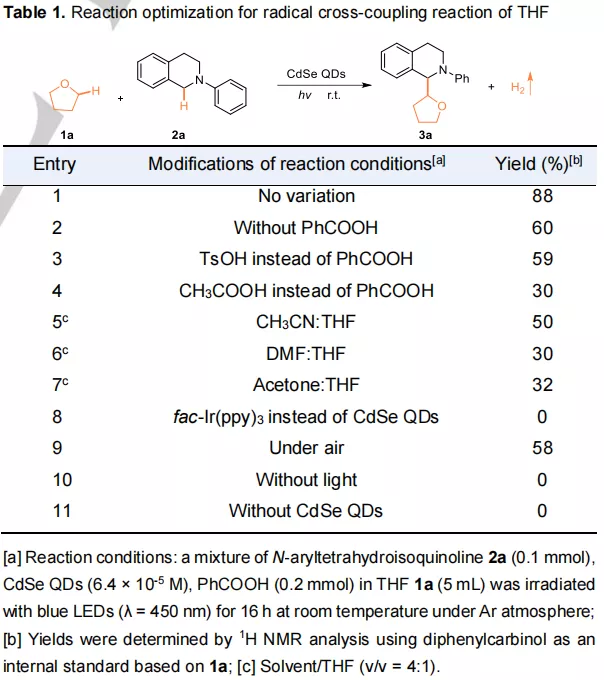
表1:THF自由基交叉偶联反应的反应优化
THF与胺的功能化得到了详尽地探究,如表1所示。只有在CdSe量子点存在下光照,才能驱动THF与胺发生交叉偶联反应,且该反应对不同的胺具有普适性。
而使用其他的分子光催化剂如 Ir(bpy)3、Ru(bpy)3等则不能催化该反应的进行。

图2:室温下Ar气氛下DMPO(1.8×10-2 M)、CdSe量子点(2.9×10-5 M)在DMF中溶液的EPR图谱:(i)黑暗;(ii)在DMF/THF中(v/v=6:1);(iii)包含2a(2.86×10-2M);(iv)含有THF和2a(2.86×10-2 M)。
如图2所示,通过控制EPR实验的条件验证了THF与胺交叉偶联的机理。
在同时加入THF与N-芳基四氢异喹啉参与反应的条件下,EPR检测到了α-胺碳中心自由基和α-氧烷基自由基的信号,即CdSe量子点能够同时活化胺和THF的C-H键,促进两者进行交叉偶联反应同时产生氢气,实现了在非常温和的条件下充分利用量子点的光生电子和光生空穴。
除此以外,作者还对量子点光催化活化THF的α-C-H键反应进行了大量的不同底物拓展实验,结果表明,该反应对不同的反应底物都具有良好的普适性,证明了量子点光催化活化THF的α-C-H键在实际合成应用中的巨大潜力。
相关成果以“Direct, Site-Selective and Redox-Neutral α-C–H Bond Functionalization of Tetrahydrofurans via Quantum Dots Photocatalysis”为题,发表在国际高水平期刊《Angewandte Chemie International Edition》上。
文章链接:
https://onlinelibrary.wiley.com/doi/abs/10.1002/anie.202109849
本文所有内容文字、图片和音视频资料,版权均属科学指南针网站所有,任何媒体、网站或个人未经本网协议授权不得以链接、转贴、截图等任何方式转载。








