









 您已经拒绝加入团体
您已经拒绝加入团体

 2025-08-21
2025-08-21
 3842
3842
 0
0
【摘要】 基于FDA/EU微生物限值要求,详解药品冷冻干燥机真空泄漏率测试的科学方法。包含空隙体积测量技术、风险时段分析及0.027mbar/30min验收标准的实践依据,确保无菌制剂生产合规性。
药品冷冻干燥机真空完整性测试的验收标准建立需基于科学方法。尽管无菌注射剂生产对冻干机密封性有严格要求,目前行业仍缺乏统一的泄漏率验收标准。值得注意的是,FDA 100级与欧盟A级洁净区的微生物限值高度近似(均要求<1 cfu/m³),但冻干机内部环境分类尚未明确定义。
核心科学原理(图1示意):
通过测量冻干系统空隙体积(腔室+冷凝器+连接管路的总空气空间),结合最坏场景假设——泄漏源位于非洁净控制的机械室内,计算生物负荷膨胀后的最大允许泄漏量。该方法确保泄漏空气进入后,系统仍满足100级/A级微生物标准。
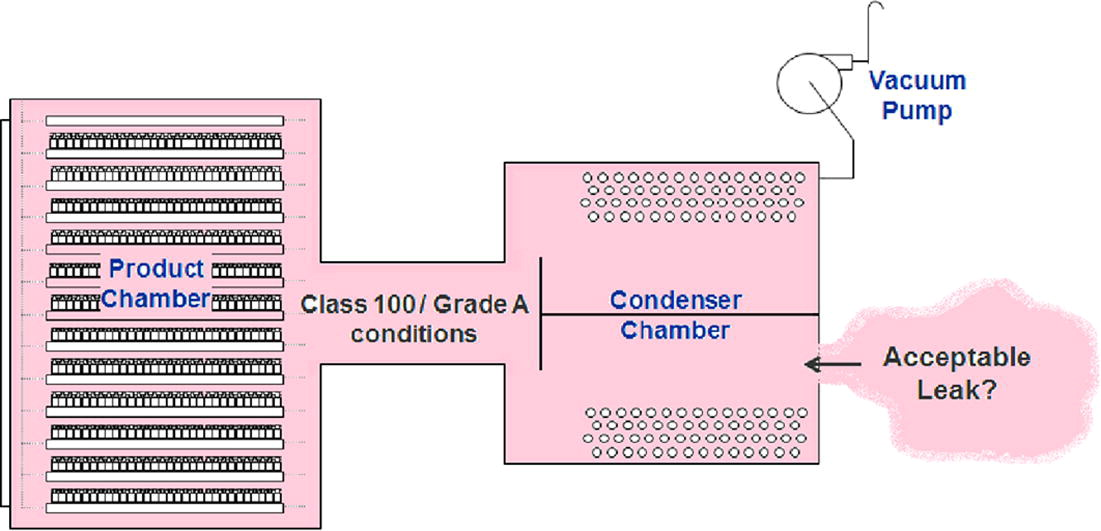
图1 药品冷冻干燥机泄漏率验收标准的科学依据。
关键风险时段验证
二次干燥全过程(从一次干燥结束至压塞完成)是微生物污染高风险期。此阶段瓶内无正压保护,且泄漏率保持稳定。需特别说明:一次干燥因升华蒸汽外排形成正压,污染风险较低。
空隙体积测量方法
采用图2所示1.18L不锈钢罐(带卫生级蝶阀),通过理想气体定律计算:
ΔP·V = Δn·R·T
(V=空隙体积;ΔP=腔室压差;Δn=注入空气摩尔数;T=机械室温度)

图2 不锈钢罐,容量1.18 L,带有内部蝶阀和卫生连接。
工业实践案例
IMA Edwards冻干机验证流程表明:
1.操作资格认证需满足ISPE真空保持标准
2.生产循环前后执行泄漏测试(30分钟压升≤0.027mbar)
3.冷凝器位于非洁净区需重点监控
该方法为建立药品冻干机真空密封验收标准提供量化依据,确保无菌制剂生产合规性。
参考文献:1.Hardwick, L. M.; Nail, S. L.; Jarman, J.; Hasler, K.; Hense, T., A proposed rationale and test methodology for establishment of acceptance criteria for vacuum integrity testing of pharmaceutical freeze dryers. Eur. J. Pharm. Biopharm. 2013, 85 (2), 236-239.
科学指南针在全国建立32个办事处和20个自营实验室,拥有价值超2.5亿元的高端仪器。检测项目达4000+项,覆盖材料测试、环境检测、生物服务、行业解决方案、模拟计算等九大业务。累计服务1800+个高校、科研院所及6000+家企业,获得了60万科研工作者的信赖。
免责声明:部分文章整合自网络,因内容庞杂无法联系到全部作者,如有侵权,请联系删除,我们会在第一时间予以答复,万分感谢。








