









 您已经拒绝加入团体
您已经拒绝加入团体

 2026-04-16
2026-04-16
 1755
1755
 0
0
【摘要】 科学指南针主办第四届多尺度模拟与材料智能计算论坛,10 位计算领域权威专家分享 AI 赋能材料计算、DFT 计算、分子模拟、跨尺度模拟、催化剂设计、锂电池研发等前沿成果,提供一站式材料计算测试服务。

行业背景:计算化学赋能材料研发革新
当前材料科学研发正朝着高效化、精准化、智能化转型,传统实验试错模式难以满足高通量筛选与跨尺度机理研究需求。计算化学、多尺度模拟与人工智能技术的深度融合,成为破解材料设计瓶颈、缩短研发周期的核心路径。
在中国化学会第 35 届学术年会期间,科学指南针・唯理计算主办第四届多尺度模拟与材料智能计算论坛,汇聚国内外 10 位计算领域权威专家,围绕 AI 赋能材料计算、多尺度模拟、高通量筛选等关键方向,分享前沿成果与应用实践,为 DFT 计算、分子模拟、材料智能设计等领域提供权威指引。
一、分子与基础化学体系计算前沿
1. 分子间相互作用与量子动力学研究

谢代前 南京大学教授
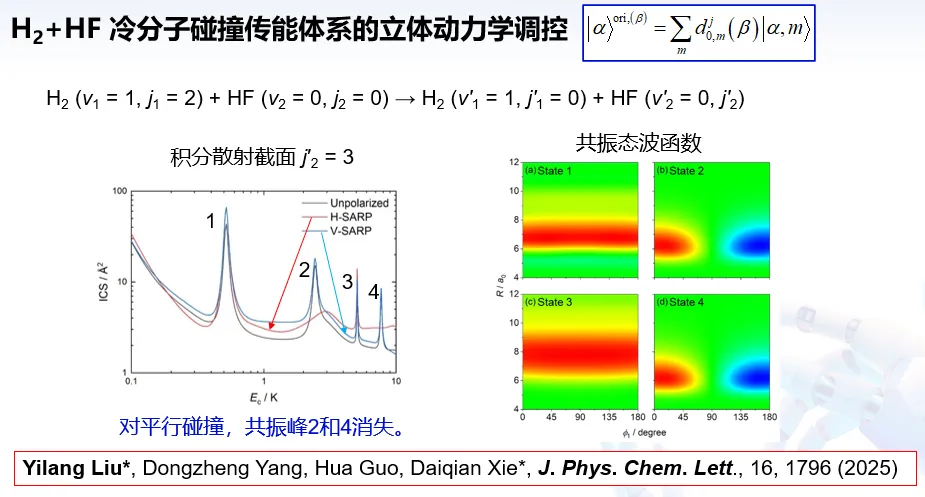
南京大学谢代前教授聚焦分子间非弹性散射与冷分子碰撞动力学,针对高精度势能面构建、量子动力学计算效率提升等难题,开发小分子体系势能面精确计算方法与非含时碰撞传能动力学新方法,揭示冷分子碰撞微观动力学机制,为分子动力学基础研究提供理论支撑。
2. 分子与框架材料的 AI 计算设计

赵纪军 华南师范大学教授
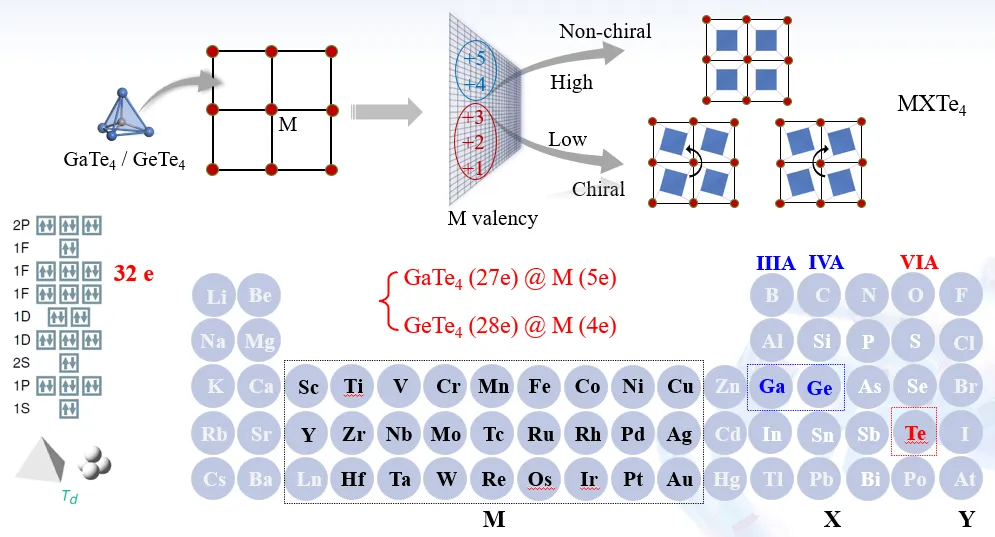
华南师范大学赵纪军教授以有机分子与金属有机框架材料为研究对象,基于深度学习图神经网络精准描述分子激发态能级,高效筛选三重态裂分特性有机分子;同时通过原子尺度调控金属有机框架的节点与桥联结构,实现功能材料理性构筑,拓展分子基功能材料研发边界。
3. 张量性质预测的几何深度学习框架

徐昕 复旦大学教授
复旦大学徐昕团队提出等变图神经网络通用输出模块,可端到端预测分子与晶体任意阶数、任意对称性张量性质,结合 XPaiNN 模型实现与第一性原理计算媲美的精度,支持原子层面性质一体化预测,为 AI 驱动的功能分子与材料发现提供通用算法框架。
二、催化与电化学材料计算创新
1. 数据驱动的电催化剂理性设计

肖建平 中国科学院大连化学物理研究所研究员
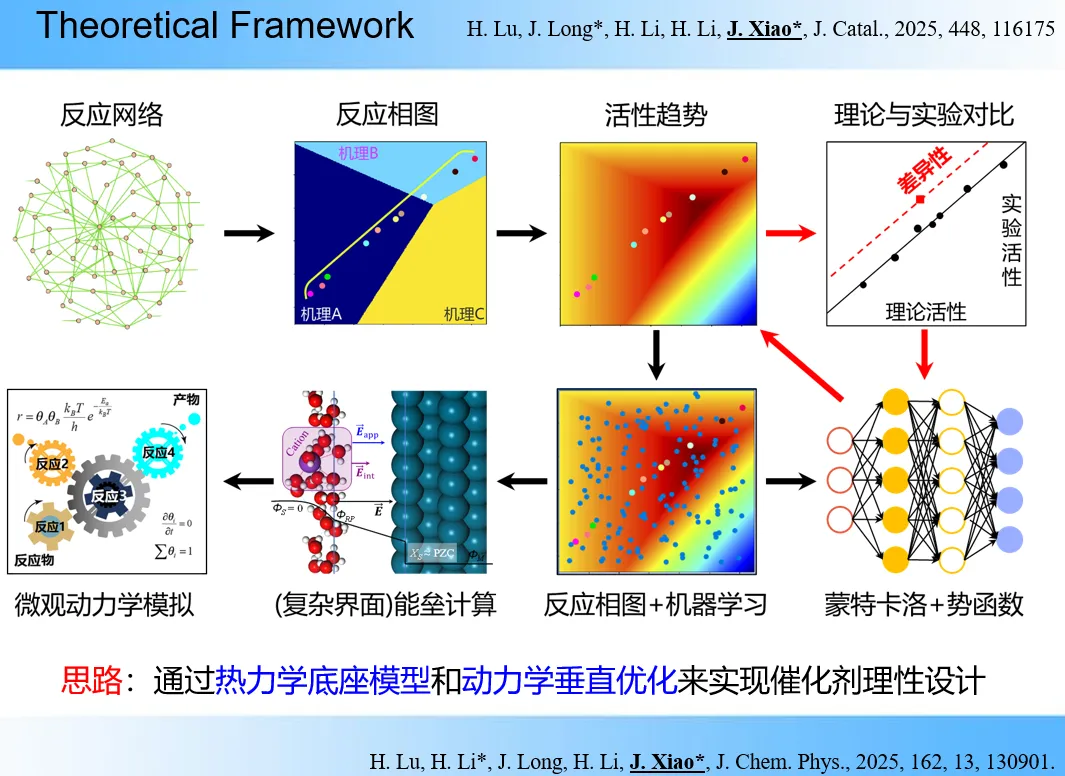
中科院大连化物所肖建平研究员开发反应相图与恒电势计算方法,揭示氮氧化物电催化转化机理,精准预测催化剂活性与选择性;基于该方法筛选出高活性 Cu6Sn5 合金催化剂,实现安培级合成氨性能,为电催化脱硝、合成氨等绿色催化技术提供设计方案。
2. 电化学界面单原子催化剂轨道调控

刘锦程 南开大学特聘研究员
南开大学刘锦程研究员通过恒电势计算,阐明电极电势对单原子催化剂 d 轨道电子占据的调控机制,发现配体修饰可激活惰性 Ni 位点;结合恒电势机器学习分子动力学,揭示碱金属阳离子与界面电场的协同作用,为单原子催化剂精准设计提供理论依据。
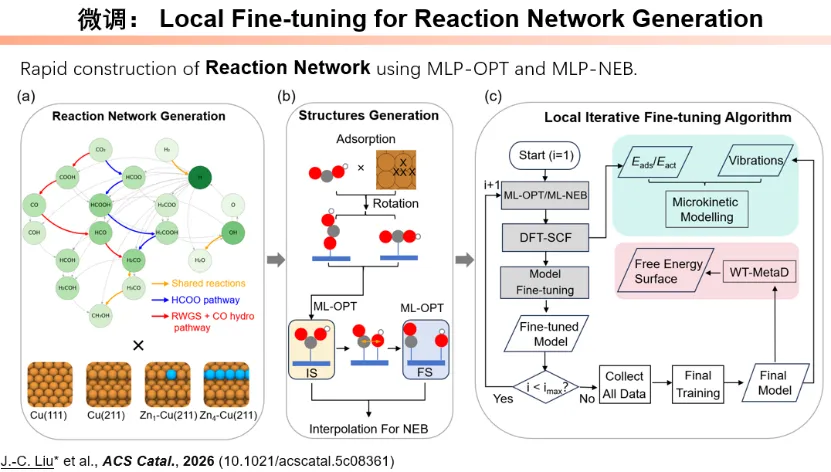
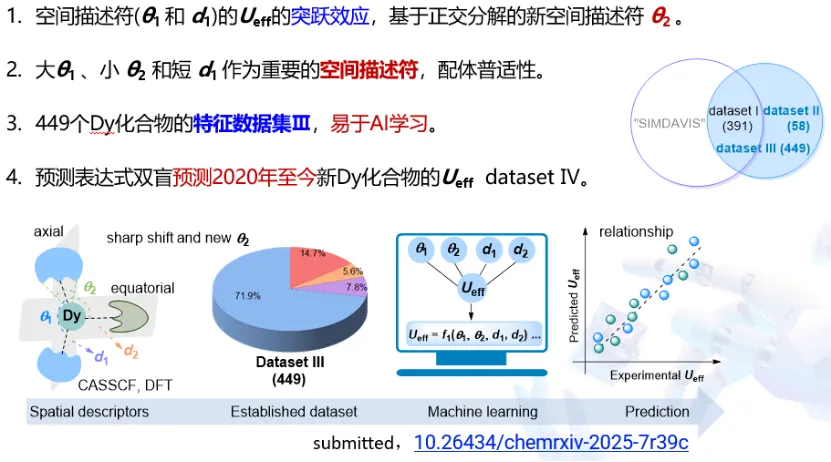
3. 稀土有机配合物反应机理与磁体预测

彭谦 南开大学教授
南开大学彭谦教授建立稀土有机壳层模型,总结三类稀土有机反应机制,揭示弱配位、中等配位、强碱性配体下的动态配位与轨道次级作用规律,突破传统 σ- 键复分解机理局限,为稀土基单分子磁体、光电磁功能材料研发提供核心理论支撑。
三、电池与固态电解质跨尺度计算
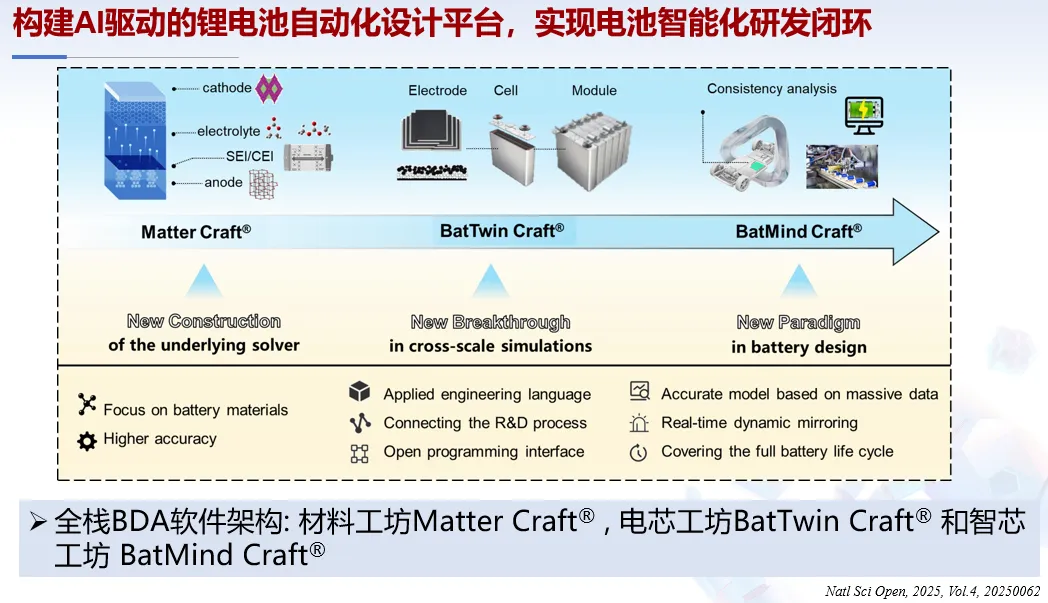
1. AI 驱动锂电池跨尺度模拟与自动化设计

郑家新 北京大学长聘副教授
北京大学郑家新副教授针对锂金属负极跨时空尺度界面行为,开发耦合电荷矩张量势与 HAML 框架,实现 SEI 组分演化动态追踪;联合企业搭建电池设计自动化(BDA)平台,融合跨尺度模拟与 AI 算法,构建从原子设计到系统预测的全流程智能化研发体系。
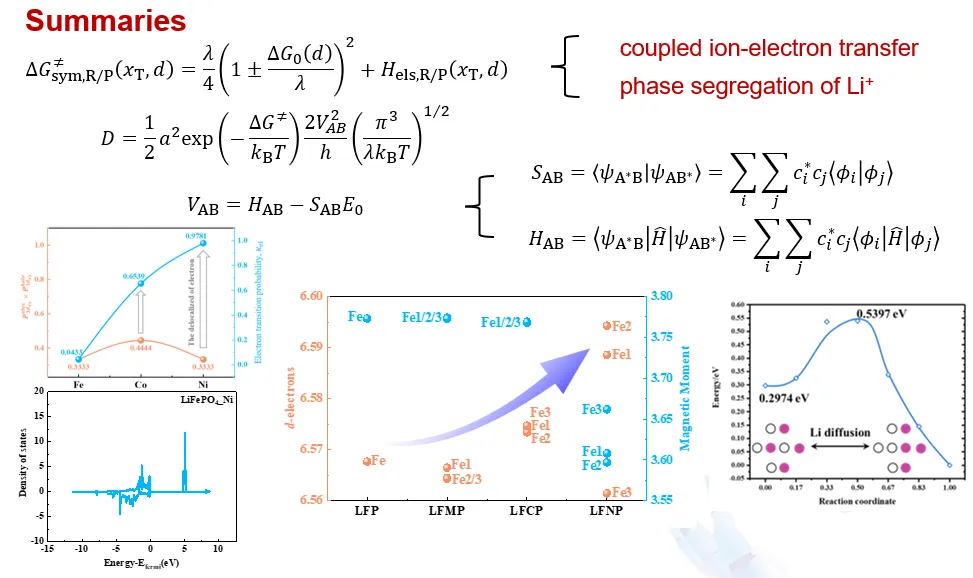
2. 磷酸铁锂锂离子传输动力学计算

陈胜利 武汉大学教授
武汉大学陈胜利教授结合密度泛函理论与神经网络深度势,阐明磷酸铁锂材料中 Li + 离子 - 电子耦合转移机制,揭示掺杂对锂离子扩散速率的调控规律,精准估算锂离子扩散系数,为提升磷酸铁锂正极倍率性能提供理论指导。
3. 固态电解质界面效应与多尺度模拟

唐法威 科学指南针·唯理计算首席工程师
科学指南针・唯理计算唐法威首席工程师聚焦固态电解质界面调控难题,通过多尺度模型与有限元模拟,揭示晶界对离子传输、锂枝晶形核的影响机制;提出非对称设计策略与纳米结构失稳热力学判据,为高性能固态电解质开发提供计算支撑。
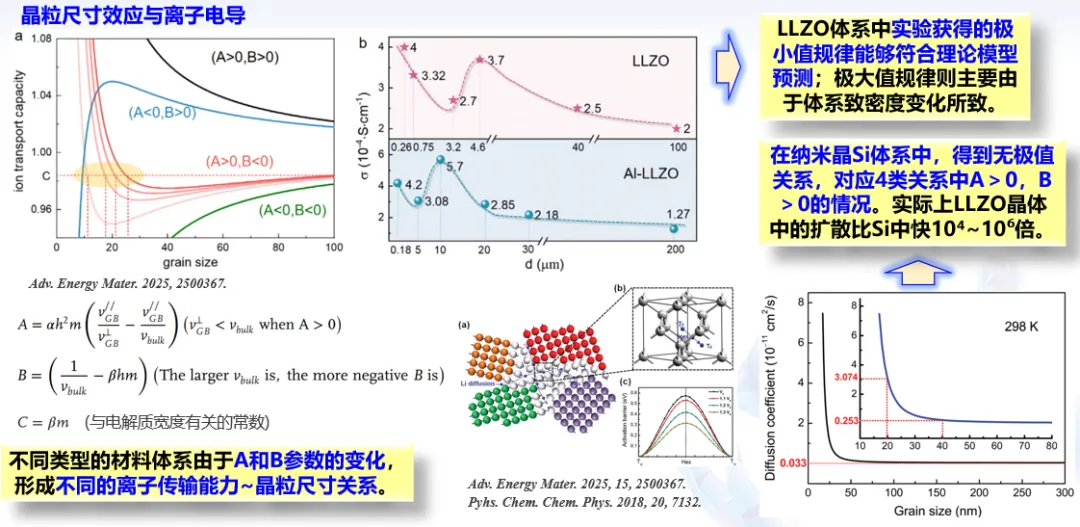
四、通用 AI 原子模拟技术突破

刘智攀 复旦大学教授
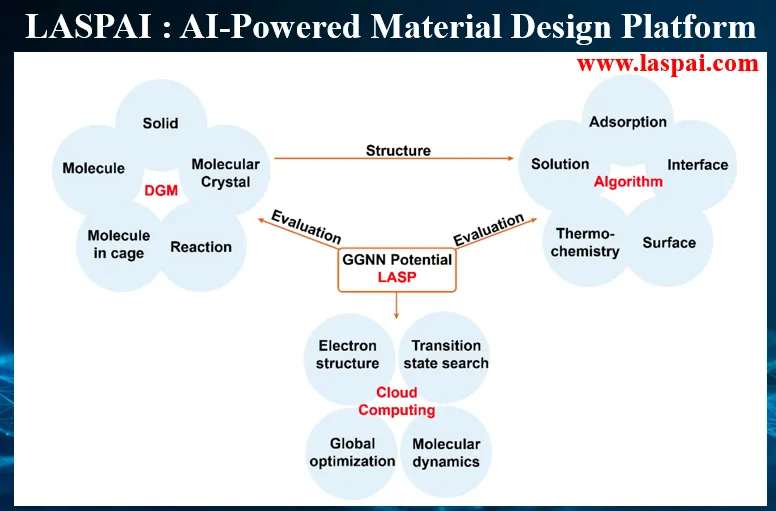
复旦大学刘智攀教授提出高阶配对简化神经网络模型,以低成本实现高表示能力势函数构建,基于 LASP 项目数据集训练全元素通用全局神经网络势能;结合通用结构生成模型,搭建 LASPAI 网页计算平台,为大规模原子模拟与材料快速预测提供通用解决方案。
应用价值:计算技术重构材料研发模式
本次论坛展示的AI + 多尺度计算技术,已从传统的实验解释工具,升级为材料理性设计核心手段。在催化、电池、固态电解质、分子材料、稀土功能材料等领域,可实现机理解析、高通量筛选、性能预测全流程覆盖,大幅降低研发成本、缩短研发周期。
科学指南针依托本次论坛前沿成果,为科研团队与企业提供 DFT 计算、分子模拟、AI 材料设计、多尺度模拟等一站式计算测试服务,助力材料科学研发数字化、智能化升级。
总结
第四届多尺度模拟与材料智能计算论坛,汇聚计算化学与 AI 材料领域顶尖智慧,覆盖分子动力学、催化设计、电池研发、固态电解质、通用原子模拟等核心方向,形成完整的AI + 材料计算技术体系。
未来,科学指南针将持续深耕材料计算模拟领域,搭建学术交流与技术服务平台,推动多尺度模拟、人工智能与材料科学深度融合,为全球材料研发与产业升级提供专业技术支撑。








