









 您已经拒绝加入团体
您已经拒绝加入团体

 2026-01-29
2026-01-29
 2005
2005
 0
0
【摘要】 本文详解吸附能计算在催化、电池、储氢等领域的物理机制,涵盖表面能、悬挂键等关键概念及VASP实操案例。科学指南针平台提供专业DFT计算服务,支持材料吸附性能研究。
吸附能计算的核心价值与物理意义
吸附能计算是密度泛函理论(DFT)在材料科学中的核心应用,通过量化分子与材料表面的相互作用能,揭示催化、储能等过程的物理机制。科学指南针平台集成VASP等专业工具,提供精准吸附能计算服务,助力顶刊研究【科学指南针·DFT模块】。吸附能计算在固体表面催化、电极材料设计、储氢材料优化等领域具有广泛应用,是连接理论与实验的关键桥梁。
物理机制:表面能与悬挂键的起源
材料表面原子因化学键“切断”形成悬挂键,导致表面能高于块体材料:
-
悬挂键定义:表面原子未饱和轨道,如Pt(001)面原子配位数降低(图1);
-
表面能计算:通过吸附能评估表面活性,指导催化剂设计;
-
亚稳特征:悬挂键使表面处于亚稳态,需通过吸附能计算优化稳定性。
科学指南针平台自动化表面模型构建,确保计算准确性【科学指南针·模型构建】。
%E8%A1%A8%E9%9D%A2%E7%9A%84%E5%BD%A2%E6%88%90%E8%BF%87%E7%A8%8B.jpg)
图1 Pt(001)表面的形成过程
典型应用领域与案例解析
吸附能计算通过量化分子-表面相互作用,推动材料性能优化:
固体表面催化(固氮、CO₂转化、ORR等)
-
机制分析:悬挂键与反应物形成化学吸附,弱化分子内键(如异丙醇OH基团与Mo作用);
-
电荷转移:差分电荷显示电子重分布,降低反应能垒(图2);
-
顶刊案例:Journal of Materials Chemistry A研究证实吸附能计算指导催化剂设计。
科学指南针平台支持多类型催化反应模拟,覆盖贵金属、单原子催化等体系。
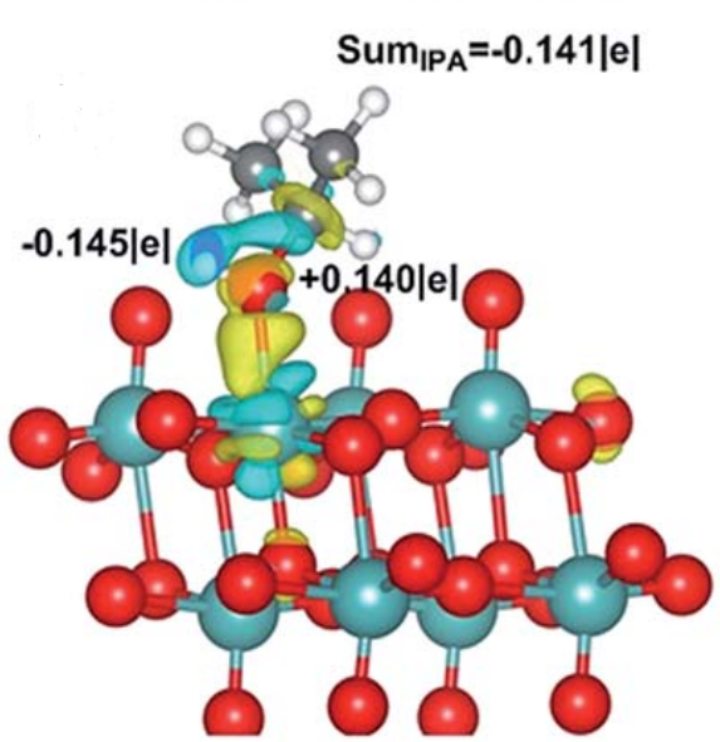
图2 异丙醇在MoO3-X上的吸附
电极材料设计(锂/钠/铝电池)
-
吸附强度平衡:太弱则容量低,太强则迁移能垒高(图6);
-
性能优化:通过吸附能计算调控离子吸附强度,提升电池容量与循环稳定性;
-
案例参考:Journal of Materials Chemistry A研究展示C₃N₂材料中锂离子吸附能与迁移能垒关联。
科学指南针平台提供电极材料吸附能-扩散协同分析工具。
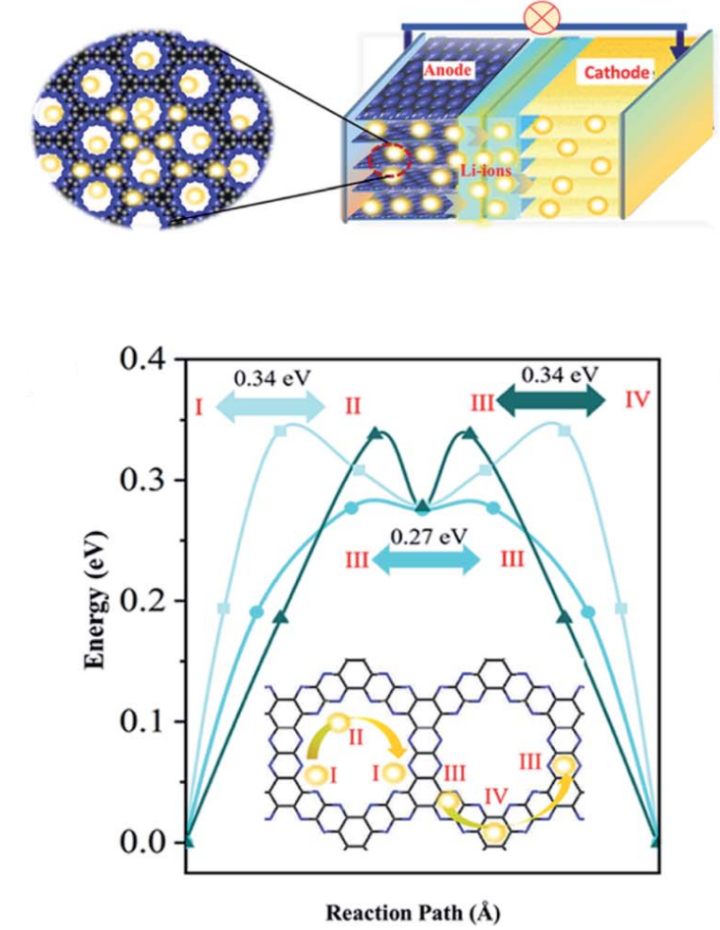
图6 锂离子在C3N2多孔材料上的吸附与迁移能垒
储氢材料性能优化
-
吸附能阈值:需平衡氢气吸附与脱附过程,避免过强吸附导致释放困难;
-
动力学模拟:结合分子动力学分析氢气捕获/释放路径;
-
研究支持:ACS Materials Letters报道吸附能计算指导储氢材料设计。
科学指南针平台集成多尺度模拟,优化储氢材料性能。
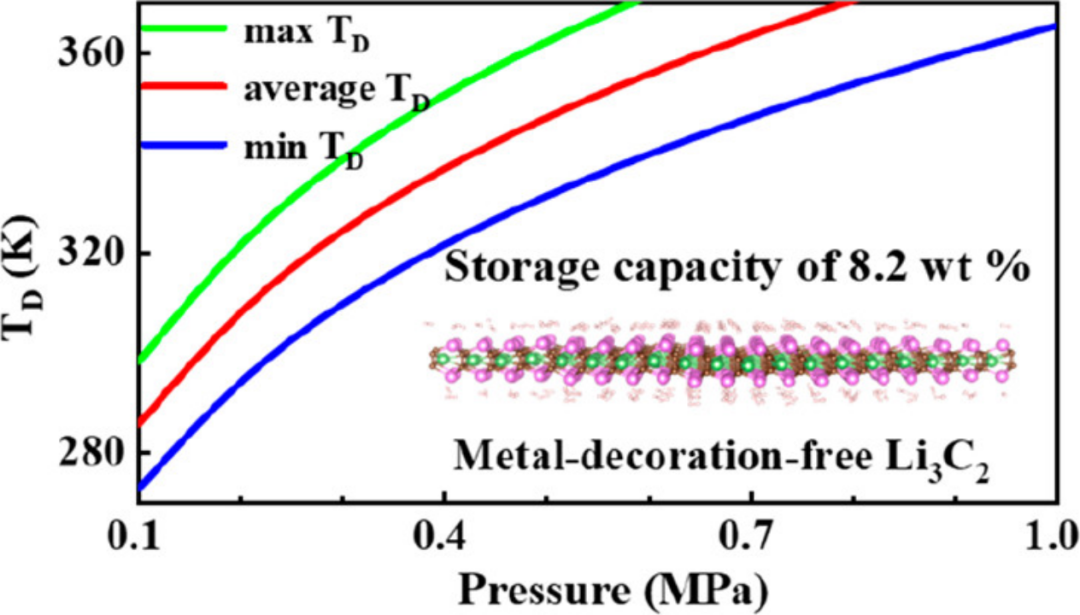
吸附能计算的最佳实践与工具
科学指南针平台推荐以下VASP吸附能计算流程:
-
模型构建:精准切割表面,定义吸附位点;
-
参数设置:优化k点采样、泛函选择(如PBE用于表面计算);
-
结果验证:对比实验数据(如Pt(111)甲醇吸附热),校准计算误差。
平台提供自动化脚本,减少手动操作错误【科学指南针·计算流程】。
结语与平台服务
吸附能计算是理解材料表面行为、优化性能的核心技术。科学指南针平台提供从模型构建到数据分析的全流程DFT计算服务,支持研究人员快速获得可靠吸附能数据。如需VASP计算或吸附能分析支持,欢迎联系科学指南针团队【科学指南针·服务咨询】。








