







 您已经拒绝加入团体
您已经拒绝加入团体

 2022-10-08
2022-10-08
 3588
3588
 0
0
【摘要】 时至今日,计算化学早已不仅仅是实验的附属品了,它随着计算水平的逐步提升和理论框架的逐步完善,现在,越来越多的纯计算可以单独发在以往的顶刊之上了。
时至今日,计算化学早已不仅仅是实验的附属品了,它随着计算水平的逐步提升和理论框架的逐步完善,现在,越来越多的纯计算可以单独发在以往的顶刊之上了。
从本期起,我们将在每周推出《一周顶刊计算报道》,它主要网罗了上一周发表在各大顶刊的纯计算文章,它包括但不限于电子结构计算、量化计算、动力学模拟、蒙特卡洛模拟以及机器学习等等。还等什么,让我们一起出发吧!
1.PNAS:固有溶剂响应的离子依赖的蛋白质表面相互作用
.webp)
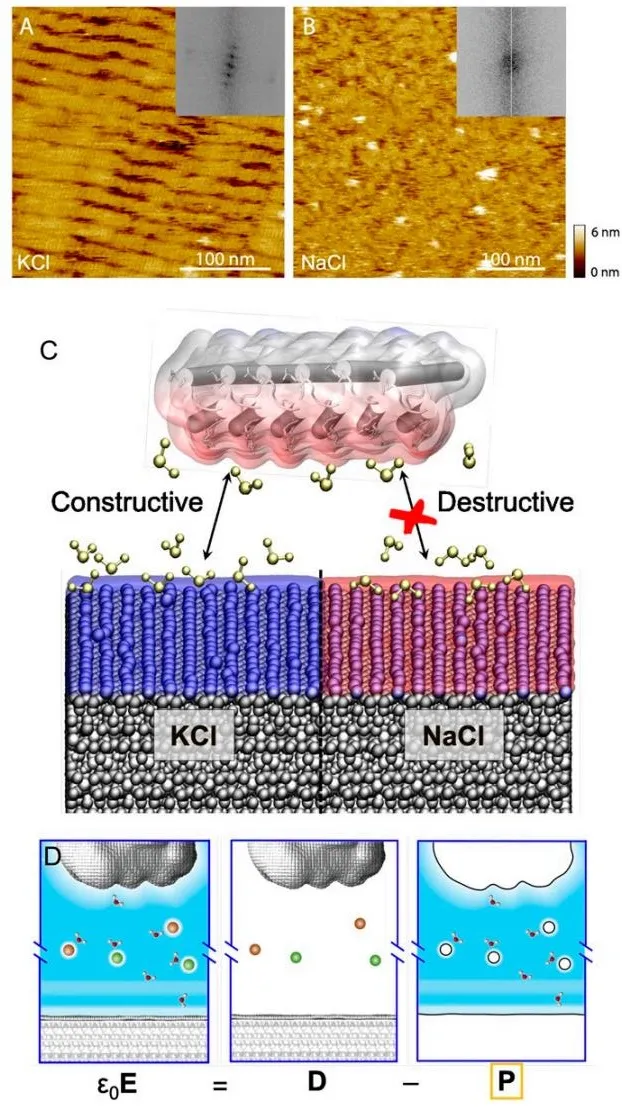
层状硅酸盐矿物白云母,可广泛用作大分子模式的表面模板,但由于缺乏对其在不同溶液条件下的表面化学的分子理解,这需要预测和控制吸附物种的自组装。在此,来自美国加州大学圣迭戈分校的F. Akif Tezcan & 罗格斯大学的F. Akif Tezcan & 华盛顿大学的Christopher J. Mundy等研究者,利用全原子分子动力学模拟与基于局部分子场理论的静电分析相结合,提供了远程和短程静电的干净分离。利用水的极化响应作为一种测量电场的方法,这些电场是由图形化的、表面束缚的离子产生的,这些离子引导着带电大分子的吸附,研究者应用不对称极化表面引起的力的Landau理论来计算两个与白云母结合的蛋白质(DHR10-mica6和C98RhuA)的表面相互作用。在高浓度的KCl和NaCl水溶液中,表面和蛋白质之间的压力比较,揭示了蛋白质远场表面相互作用的离子特异性差异,巧妙地捕获离子调节白云母表面电荷的能力,进而选择性地吸引每个蛋白质的一个结合面。
参考文献:
Jesse L. Prelesnik, Robert G. Alberstein, Shuai Zhang, Harley Pyles, David Baker, Jim Pfaendtner, James J. De Yoreo, F. Akif Tezcan, Richard C. Remsing, Christopher J. Mundy. Ion-dependent protein surface interactions from intrinsic solvent response. Proceedings of the National Academy of Sciences Jun 2021, 118 (26) e2025121118; DOI: 10.1073/pnas.2025121118
原文链接:
https://www.pnas.org/content/118/26/e2025121118
2.Nature Materials:三维网状聚合物分子构型的计算预测
.webp)
天然和合成网络材料的三维排列方式,决定了其应用范围。实时控制每个构件和官能团的结合,是为了从分子水平调节材料的宏观特性。在此,来自美国斯坦福大学的Reinhold H. Dauskardt & 比利时根特大学的Reinhold H. Dauskardt等研究者,报告了一种结合动力学蒙特卡罗和分子动力学模拟的方法,该方法可以从化学和物理上预测三维网络的整个形成过程中,积木在时间和空间上的相互作用。该理论框架考虑了分子间和分子内化学反应活性、扩散率、节段组成、分支/网络点位置和缺陷的变化。
从动力学和三维结构信息收集,研究者构建了结构-性质关系的分子描述符,如孔隙大小或悬浮链分布,并区分理想和非理想结构元素。研究者通过合成具有定制性质和功能的有机硅、环氧胺和Diels-Alder网络验证了以上关系,进一步证明了该平台的广泛适用性。
.webp)
参考文献:
De Keer, L., Kilic, K.I., Van Steenberge, P.H.M. et al. Computational prediction of the molecular configuration of three-dimensional network polymers. Nat. Mater. (2021). https://doi.org/10.1038/s41563-021-01040-0
原文链接:
https://www.nature.com/articles/s41563-021-01040-0#citeas
3.ACS Catalysis:弱催化线粒体伴侣Trap1机制的原子模拟:结构不对称对反应活性影响的洞见
.webp)
线粒体伴侣Trap1是一种ATP酶蛋白,负责监督客体蛋白的正确折叠,并在某些癌症中表现出活性和/或表达水平的改变。通过诱导广泛的结构重排,ATP的裂解是Trap1完成其任务的必要条件;同时,Trap1缓慢的ATP转换是调节其构象周期和介导其的残基间串扰信号的控制开关之一。
值得注意的是,Trap1的水解需要采用一种独特的不对称封闭的同二聚体构象,其中与弯曲的前聚体(Buc)结合的ATP优先被裂解,而与直的前聚体(Str)的水解仍然不受青睐。然而,由于其内在机制的复杂性和其前聚物活性位点的相似性,Trap1的不对称性和其特征反应性之间的分子联系仍然难以捉摸。为了解决这个问题,来自意大利帕维亚大学的Giorgio Colombo等研究者,在此报告了Trap1潜在ATP酶机制详细的计算研究。通过经典分子动力学(MD)模拟,研究者监测了ATP和亲核水WatNuc在Buc和Str中获得反应姿态的频率。
半经验杂化量子力学-分子力学(QM/MM) MD模拟结合伞式采样和密度泛函理论计算基准,然后用于对两个可能的水解途径的每个前聚体内的反应自由能垒进行采样。酶辅助水解,具有类似偏磷酸盐的过渡状态和催化谷氨酸脱质子WatNuc,发现在两个前聚物中比底物辅助水解更受青睐。然而,研究者发现在Buc中,WatNuc被另一个水分子WatTyr与附近的酪氨酸结合的隔离要罕见得多,这证明了这种罕见的隔离降低了酶辅助途径的反应能垒。因此,研究者确定(生物学上重要的)酪氨酸是在Buc中有利于ATP水解的主要中介,而不是Str。这里改进的Trap1反应性模型在实验和结构上是一致的,可进一步帮助开发蛋白质活性的选择性调节剂。
.webp)
参考文献:
Stefano A. Serapian, Elisabetta Moroni, Mariarosaria Ferraro, and Giorgio Colombo. Atomistic Simulations of the Mechanisms of the Poorly Catalytic Mitochondrial Chaperone Trap1: Insights into the Effects of Structural Asymmetry on Reactivity. ACS Catalysis 0, 11 DOI: 10.1021/acscatal.1c00692
原文链接:
https://pubs.acs.org/doi/10.1021/acscatal.1c00692
4.ACS Catalysis:枚举金属纳米粒子上的活性位点:理解钴粒子大小对CO解离的依赖
.webp)
对结构敏感性的详细了解,是多相催化的中心主题,对指导改进催化剂的合成很重要。遗憾的是,人类目前无法准确地列举出普遍存在的金属纳米颗粒催化剂上的特定活性位点,从而阻碍了催化剂研究的进展。
在此,来自荷兰埃因霍温科技大学的Ivo A. W. Filot等研究者,利用量子化学数据训练力场的原子模拟,对钴粒子的形状作为其大小的函数进行了取样。基于模式识别的算法用于识别与CO离解相关的表面原子排列,而CO离解是费托(FT)反应的关键步骤。在1-6 nm范围内,较大的纳米粒子能够催化C-O键的低势垒断裂的阶梯边位数明显增加。结合FT反应的微观动力学,研究者可以重现实验FT活性趋势。阶梯边缘位点的稳定性与平台纳米岛在较大纳米颗粒上的稳定性增加有关。
.webp)
参考文献:
Michel P. C. van Etten, Bart Zijlstra, Emiel J. M. Hensen, and Ivo A. W. Filot. Enumerating Active Sites on Metal Nanoparticles: Understanding the Size Dependence of Cobalt Particles for CO Dissociation. ACS Catalysis 0, 11 DOI: 10.1021/acscatal.1c00651
原文链接:
https://pubs.acs.org/doi/10.1021/acscatal.1c00651
5.ACS NANO:通过高光谱扫描隧道光谱数据的机器学习揭示Adatom阵列中的化学键
.webp)
表面上的吸附原子阵列提供了一个理想的平台,可通过改变局部电子隧穿光谱来探索化学键的机制。虽然这些信息在高光谱扫描隧道光谱数据中很容易获得,但由于缺乏合适的分析工具,其分析受到了很大的阻碍。
在此,来自美国橡树林国家实验室的Zheng Gai & Sergei V. Kalinin等研究者,开发了一种基于机器学习的工作流程,将空间域的监督特征识别和能量域的非监督聚类相结合,以揭示Co3Sn2S2剪切表面吸附原子(adatom)阵列中电子结构依赖的变化细节。这种方法,同时结合第一性原理计算,提供了使用人工神经网络来检测adatom和分类每个adatom基于他们的局部邻域组成的其他adatom。这些结构上分类的原子进一步进行频谱解卷积。利用这种方法揭示了类似构型的adatom之间电子结构的非均匀性,这表明Co3Sn2S2表面上不存在单一的原子种类,而是多种类型的原子。通过adatom地形图像的细微对比差异(或大小的轻微变化),进一步证明了上述观点。
.webp)
参考文献:
Kevin M. Roccapriore, Qiang Zou, Lizhi Zhang, Rui Xue, Jiaqiang Yan, Maxim Ziatdinov, Mingming Fu, David G. Mandrus, Mina Yoon, Bobby G. Sumpter, Zheng Gai, and Sergei V. Kalinin. Revealing the Chemical Bonding in Adatom Arrays via Machine Learning of Hyperspectral Scanning Tunneling Spectroscopy Data. ACS Nano Article ASAP DOI: 10.1021/acsnano.1c02902
原文链接:
https://pubs.acs.org/doi/10.1021/acsnano.1c02902
本文所有内容文字、图片和音视频资料,版权均属科学指南针网站所有,任何媒体、网站或个人未经本网协议授权不得以链接、转贴、截图等任何方式转载。









