









 您已经拒绝加入团体
您已经拒绝加入团体

 2021-07-26
2021-07-26
 11311
11311
 0
0
【摘要】 在实际科学研究中,分子模拟已经成为了一种从微观尺度(原子/分子)探究物质局域结构与计算宏观物性的常用手段。在进行分子模拟之前,我们首先要把相应的实际物理模型转化为分子模拟可用的结构文件,这个过程一般我们称之为“建模”。
在实际科学研究中,分子模拟已经成为了一种从微观尺度(原子/分子)探究物质局域结构与计算宏观物性的常用手段。在进行分子模拟之前,我们首先要把相应的实际物理模型转化为分子模拟可用的结构文件,这个过程一般我们称之为“建模”。
不同的分子模拟软件输入结构文件各异,比如Lammps软件的输入结构文件为data.***,Gromacs软件的输入结构文件为***.gro和***.top等。今天我们就来介绍几种常用的建模方法。
01编辑结构文件
对于一些简单体系,根据晶胞参数、原子坐标手动编辑结构文件。此方法需要对使用的分子模拟软件输入结构文件格式比较熟悉,否则容易出错,故门槛较高,但胜在灵活。
这里以Lammps软件为例建立MgO模型,输入结构文件内容如下:
.webp)
图中采用full style,详见:
https://docs.lammps.org/read_data.html的Format of a data file部分。如果想模拟更大的体系,可以在in文件中通过replicate命令实现。
02下载结构文件
直接从唯理计算小分子数据库中下载相应的cif、pdb、mol等结构文件,然后通过软件进行格式转换。
这里还是以MgO为例,从Materials Project网站:
https://materialsproject.org/#search/materials
下载MgO的Fmm空间群的cif文件。
可以通过MS、VMD、Open Babel等多种软件进行格式转换,
-
比如把cif文件导入MS中,通过Focite模块分配好电荷和力场,然后使用lammps的msi2lmp.exe进行格式转换;
-
先将cif转为pdb文件,导入VMD中,在TK console中使用pbc box显示盒子,再通过topo writelammpsdata <filename> [<atomstyle>]即可得到data文件;
-
Open Babel支持多种格式互转,官网给了几个例子:
http://openbabel.org/docs/current/Commandline_tools/babel.html#examples,
更多支持格式见以下网址:
http://openbabel.org/docs/current/FileFormats/Overview.html#file-formats。
03编辑结构文件
一些复杂体系(比如溶剂模型等),无法手动编辑亦无法采用MS手动逐个添加溶剂分子,这种情况需借助一些软件(比如packmol)。
这里以MgO水溶剂模型为例:首先准备MgO.pdb和H2O.pdb,然后准备packmol脚本***.inp,内容如下:
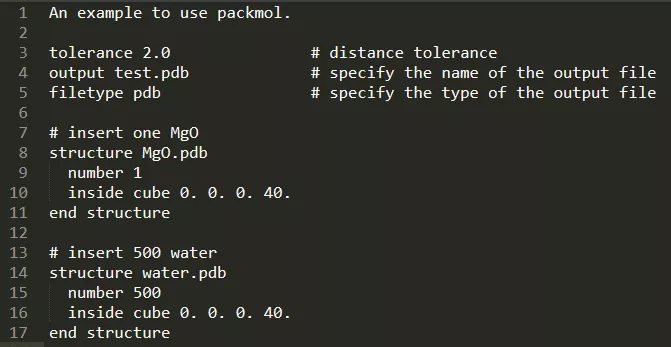
然后运行packmol < ***.inp。这里我们建立了一个含1个MgO、500个H2O的水溶剂模型,当然,在分子模拟的开始阶段别忘了先进行能量最小化。
04脚本自动建模
采用python等语言写脚本自动建模(此处可报名Python进阶小班课程,具体报名方法见详见文章)此方法仅适合高端玩家,需对各空间族群和各软件输入结构文件了然于心,不建议入门者触碰。如果您能做到这一点,恭喜您是这方面的专家!
本文所有内容文字、图片和音视频资料,版权均属科学指南针网站所有,任何媒体、网站或个人未经本网协议授权不得以链接、转贴、截图等任何方式转载。








