









 您已经拒绝加入团体
您已经拒绝加入团体

 2025-07-08
2025-07-08
 2938
2938
 0
0
【摘要】 本次会议于7月6日在厦门圆满落幕,由科学指南针·唯理计算团队主办,汇集了国内外百余位权威专家学者。会议深入探讨了计算化学在材料科学领域的创新应用,包括算法与数据融合、机器学习辅助设计、多尺度模拟突破等,聚焦能源材料、光学功能材料、电子器件、催化剂及电池技术等热点方向,总结了12位专家的前沿研究成果,为计算材料学发展提供了新思路。

7月6日,由科学指南针·唯理计算团队主办的“第三届多尺度材料计算模拟国际研讨会议”在厦门圆满落幕。来自国内外百余位权威专家学者共聚一堂,从前沿技术新应用、能源材料多尺度研究、大尺度模拟新突破等维度,深入探讨计算化学在不同材料领域应用的最新理论与研究成果,带来了一场精彩纷呈的学术盛宴。

计算论坛干货
扫码加微
领取会议回放资料
下面,小编将采撷与会专家学者的精彩观点,以飨读者:
大会报告观点集锦
| 算法-数据-知识互利共生赋能电化学储能材料创制与评价

施思齐 上海大学教授
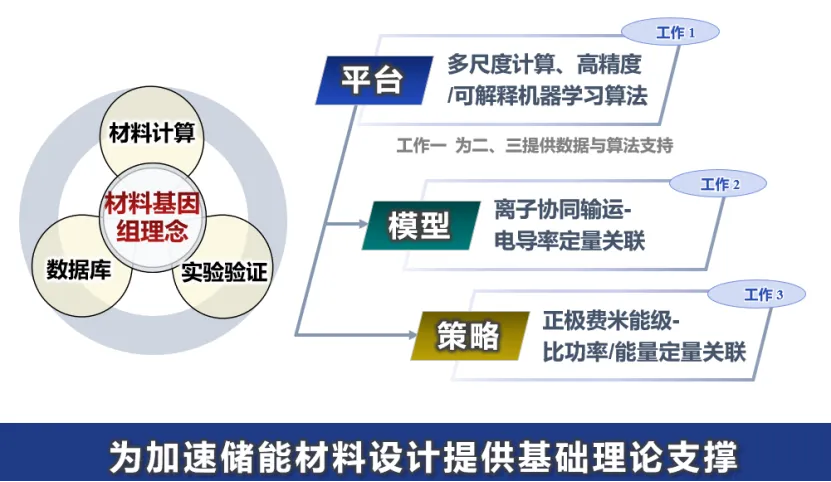
电化学储能体系的关键是材料。传统方法从材料的发现到应用往往需要十几年,很难满足产业界对更高性能电化学储能体系的要求。因此,迫切期望从学科交叉的角度,有机地融合理论、计算和实验方法,理解电池材料中电子/离子输运协同调控、界面电化学稳定性等关键科学问题,加快新材料和新体系的研发速度。
基于以上原因,施老师提出算法-数据-知识互利共生的储能材料设计范式,研究微观机制与寿命、比功率和比能量等宏观性能间的构效关系;着重结合本人和合作伙伴开展的一些工作介绍近些年来在这方面的主要进展,并对该范式在电化学储能新材料和新体系研发中扮演的角色进行了展望。
| 低维光学功能材料的计算与设计
赵纪军 华南师范大学教授
光与材料的相互作用是一个复杂而多样的过程,涉及电磁波与材料内部的量子态和集体激发的相互作用。相比其他物理化学性质,材料光学性质的第一性原理模拟计算更具挑战性。赵纪军老师的研究团队利用Gaussian、VASP等程序,基于量子限域效应、元素掺杂、官能团修饰、结构设计(多聚体、Janus、异质结)等调控策略,设计了一系列低维光学功能材料,并对相关研究内容进行了系统全面的总结。主要包括:配体金团簇和富勒烯的荧光调控;手性硒化镉量子点的光活性;二维单层材料及异质结的非线性光学性质;二维反铁磁材料 Fe2CX2(X = F, Cl)的磁光克尔效应。赵老师基于上述研究,掺杂调控了[M@Au12(dppe)5Cl2]x-2团簇的荧光波长、增强荧光效率。通过手性分子修饰CdSe量子点,引起光活性。设计了Janus结构打破二维材料的镜像对称性,引入层内偶极子,从而增强面外SHG响应、诱发内秉磁光克尔效应。上述成果为低维光学功能材料的研发提供了新的思路。

| large scale atomistic simulations of electronic devices

汪林望 中国科学院半导体研究所首席科学家
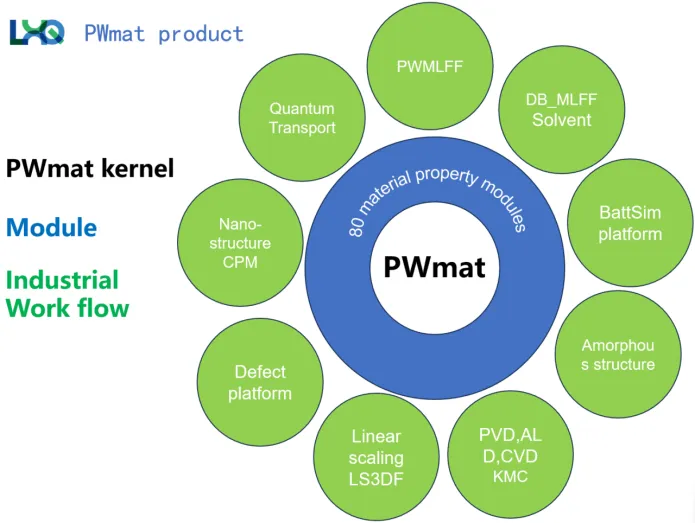
算法与计算工具的革新极大程度的影响了计算材料学与新材料设计的进程。尤其随着电子器件的尺寸缩小到纳米尺度,则需要考虑在原子尺度上模拟器件的行为。
针对上述计算痛点与焦点,汪老师系统介绍了自研软件(PWmat)的大尺度计算方法、上万原子的纳米器件模拟、表面生长模拟等内容。汪老师的团队开发了新的电荷补片方法,可用于描述原子介电屏蔽以获得器件的C-V特性。对于量子输运计算,汪老师提出了一种分治方法来构造散射态。据此能够计算出大于1万个原子器件的I-V曲线。实现了真实纳米尺度下的微电子器件模拟。此外,面对半导体日益复杂的工艺与性能标准,对相关模拟计算也提出了更具挑战性的要求。汪老师主导开发了一种快速产生机器学习力场的平台(MatPL),能够支持大尺度量子输运计算。结合上述研究成果,为电子器件的大尺度模拟提供了新的理论支撑与技术工具。
大尺度非绝热动力学理论与复杂材料载流子动力学机制

王林军 浙江大学长聘教授
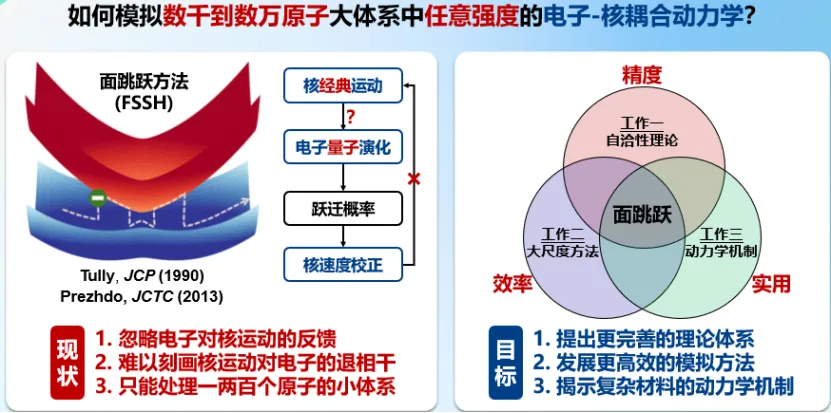
大尺度非绝热动力学涉及成千上万原子和大量电子能级,需要刻画强烈耦合的电子与原子核运动,是理论化学的重大挑战。对此,王老师提出了细致内在自洽原理,建立了自洽的混合量子-经典动力学理论,正确刻画了非绝热耦合局域时的量子退相干,在大量标准测试体系中系统提高了非绝热动力学模拟的精度;发展了不依赖非绝热耦合的非绝热动力学方法,通过电子哈密顿矩阵和退相干时间的机器学习,大幅提升了大尺度非绝热动力学模拟的效率。
进一步地,王老师团队开发了通用模拟软件,成功研究了常用方法和软件无法处理的复杂材料大体系动力学问题,实现了模型体系中任意电子耦合下百万级电子能级的载流子动力学模拟。揭示了量子点跨大势垒的反常界面电子转移机制和非原位氧化动力学机制,通过材料设计促进量子点绿光和蓝光电致发光效率首次达到理论极限和稳定空穴掺杂。
人工智能辅助的材料设计与模拟

谢禹 吉林大学教授
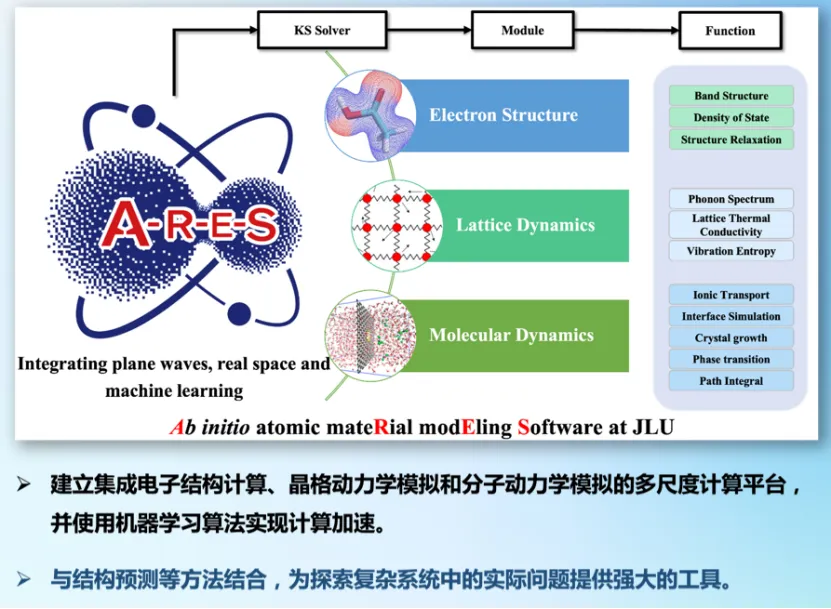
在高压、高温等非标准条件下,材料的结构稳定性与物理性质呈现出复杂的依赖关系,这对理论建模提出了严峻挑战。为应对这一问题,谢老师团队开发了第一性原理材料模拟软件 ARES。该软件基于实空间有限差分与平面波方法,具有高精度与优异的并行扩展性,能够高效执行复杂体系的电子结构计算、结构优化及分子动力学模拟任务。为了提升材料预测能力,ARES 集成了机器学习模块,支持基于主动学习的结构搜索与高精度机器学习势函数构建。在确保计算精度的前提下,大幅降低了大规模结构模拟的成本。通过高通量结构搜索与机器学习加速的势能面建模相结合,ARES 成功识别并预测了多种高压稳定相及其相关性质。尤其在多元复杂体系中,ARES 展现出对潜在稳定结构的高效发现能力。目前,ARES 已广泛应用于氧化物、硼化物、氢化物等材料体系,在理论预测与实验验证之间架起了重要桥梁,为新材料的高效发现与性能调控提供了强有力的技术支撑。
基于多尺度模拟的氧还原催化剂及其反应界面研究

李莉 重庆大学教授
燃料电池作为氢能高效转化的核心装置,其商业化进程受制于阴极氧还原反应的动力学迟缓,开发高效阴极催化电极成为突破燃料电池性能瓶颈的关键。碳基催化剂作为最有潜力的替铂催化剂,面临着本征活性根源不清、活性位密度不足、工况活性急速衰减等问题。
基于上述背景,李老师分别针对低铂催化剂和非贵碳基催化剂,采用密度泛函理论计算、分子动力学、粗粒化模拟等多尺度模拟,结合微观分子动力学方法,展开反应机理解析、催化剂设计、三相界面构筑等方面的研究。针对低铂催化剂,重点阐述了多晶面交错结构和晶面层数对反应机理、催化剂电子结构以及催化活性的影响规律;三相界面离聚物对铂基本征活性、质子通道与反应通道以及催化剂利用率的影响机制。针对碳基催化剂,重点解析了杂原子掺杂对本征活性的影响规律,多结构因素对活性位密度调控,以及失活机理等。为新型高性能燃料电池催化剂材料研发提供了理论支撑。
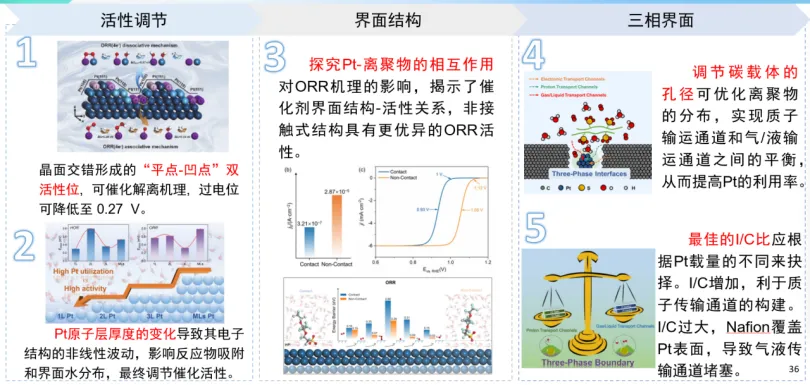
氢、氧电催化的界面结构效应

陈胜利 武汉大学教授
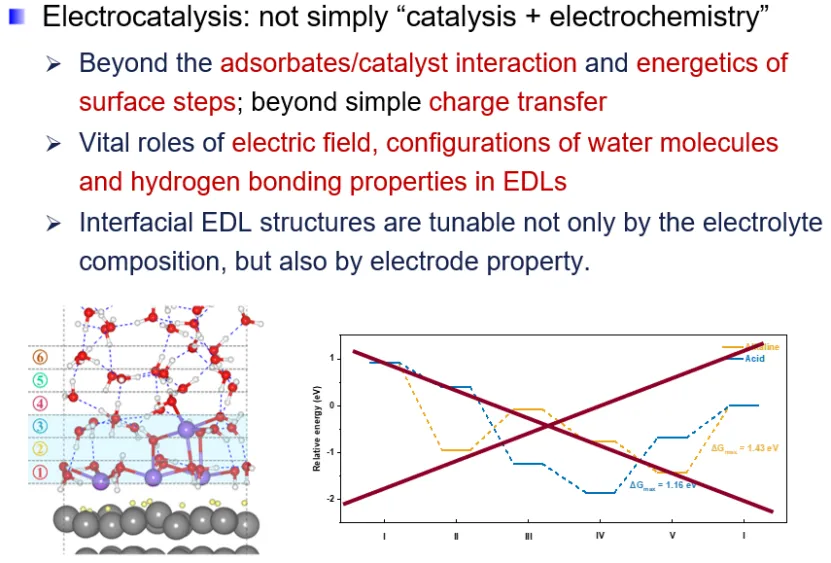
电催化反应发生在催化剂/电解质界面处,其效率同时受催化剂表面性质和界面微环境及结构的影响。以往电催化材料设计和机制研究主要聚焦于催化剂组成和结构、电解质组成等调控催化剂与反应中间体的相互作用方面。
陈老师介绍了本组最近在氢、氧电催化体系界面结构效应方面的研究结果。在氧电催化方面,针对Pt基催化剂氧还原反应高起始过电位和缓慢动力学的问题,结合实验与模拟,发现在高电位下电极表面会形成疏水的有序结构,阻碍了界面质子耦合电子转移反应的发生。并进一步证明了合金化或高指数晶面等催化剂结构组成及形貌调控策略可以通过调节电极表面疏水结构、正电荷密度进而改善界面水层结构。在氢电催化方面,结合AIMD模拟、原位表面增强红外谱学和计算谱学,揭示了溶液pH、阳离子种类、结合能等影响Pt基催化剂上氢电催化反应活性的界面机制。发现其呈现出随pH先减缓后上升的特点,即“催化剂活性-电解质pH”关系曲线存在拐点。上述研究为催化机理探索提供了新的理论支撑。
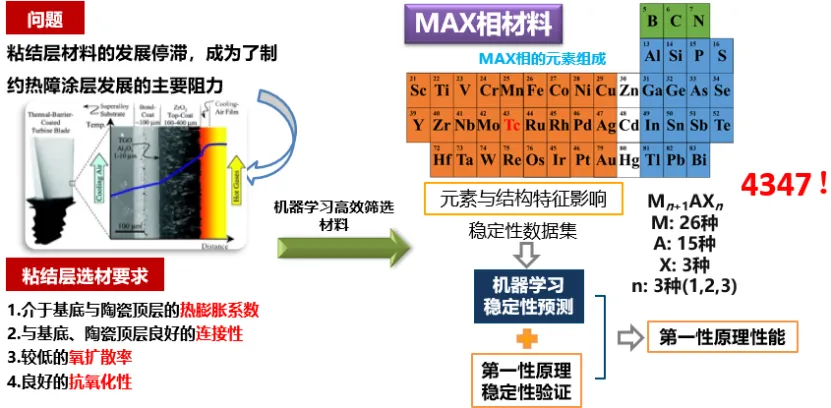
机器学习辅助陶瓷材料评价和筛选设计

柏跃磊 哈尔滨工业大学教授
机器学习可与现有理论模拟方法结合用于陶瓷材料的评价和筛选,例如连续纤维增强陶瓷基复合材料和三元层状过渡金属化合物MAX(MAB)相。柏老师采用断裂相场法生成数据训练出一个深度学习模型实现了对SiC纤维增强SiC陶瓷复合材料裂纹扩展路径的快速高精度预测,计算速度提升1-2个数量级。而基于第一性原理计算和文献报道的实验结果,采用机器学习模型分析环障涂层材料稀土硅酸盐的热膨胀系数和热导率,定量化显示了化学组成和微观结构参数的影响,实现了热膨胀系数和热导率的精确预测。进一步的,柏老师采用文献报道结果训练出一个机器学习模型用于分析和预测MAX相的物相稳定性,仅根据元素组成即可实现对稳定性的快速预测,经第一性原理确认后筛选出一系列新型化合物并成功合成出新型Ti2SnN。
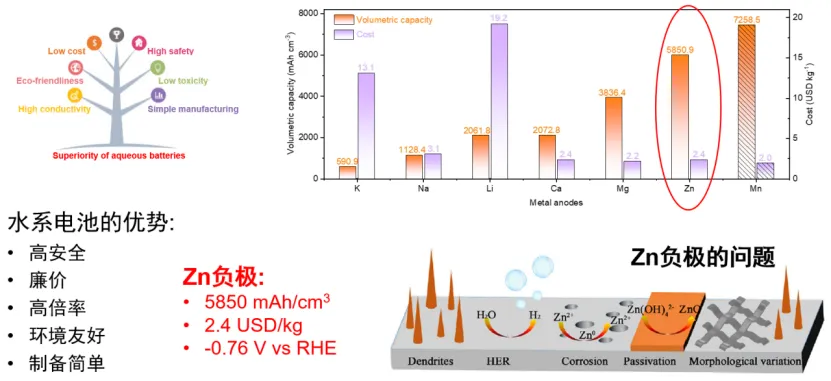
锌离子电池实用化探究

陈维 中国科学技术大学教授
双碳背景下电池起到了至关重要的作用。随着大规模储能技术的发展,Zn离子电池以高安全、低成本、高倍率、环境友好和制备简单受到了研究者的广泛关注。但是,Zn离子电池负极关于枝晶生长、界面副反应、浓差极化、力学稳定性、扩散动力学等系列问题仍制约其进一步产业化。
基于上述背景,陈老师进行了系统全面的研究,提出电极和电解液调控实现高利用率Zn负极的策略。通过电极界面调控实现高效的Zn沉积和溶解。通过SEI调控实现超高锌利用率。另一方面,通过正极调控开发新型水系锌溴固体电池,并将电解液动态稳定剂用于水系锌溴固体电池,使全电池能量密度和循环寿命显著增加。上述研究为开发大规模储能用新型电池提供了新的思路。
基于机器学习势函数的负载金催化剂理论研究

刘锦程 南开大学特聘研究员
烧结是负载金属纳米颗粒催化剂中最重要的失活方式之一。因此,了解载体对烧结行为的影响至关重要。刘老师通过原位球差校正透射电子显微镜和深度势能分子动力学计算模拟,揭示了金纳米颗粒和各种载体之间的原子尺度动态相互作用。研究发现,与非晶态二氧化硅上的金纳米颗粒相比,氧化铈上的金纳米颗粒具有更小的接触角,并且在表面台阶处的流动性明显较小。与液滴亲水性的概念类似,刘老师将小纳米颗粒的烧结迁移归因于亲金属性,它决定了金属与载体之间的相互作用。从头算分子动力学和基于机器学习的深度势能分子动力学模拟直接观察到二氧化硅表面的烧结过程和金在氧化铈上的强结合作用,并揭示了负载纳米金催化剂的尺寸效应本质,为该类催化剂的研发提供了新的理论支撑。
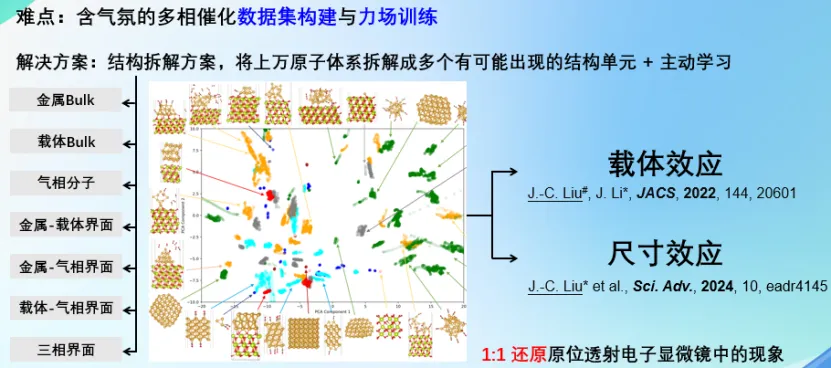
材料张量数据库与NEP自动化训练工具的开发及应用

唐刚 北京理工大学副教授
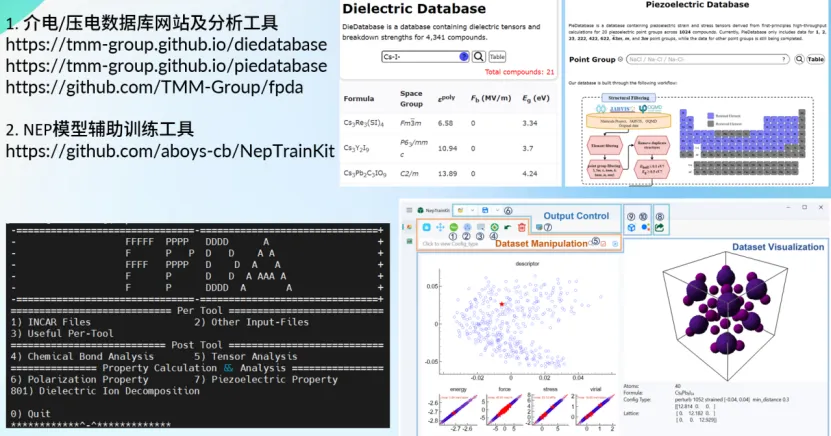
随着AI for Science研究范式的兴起,机器学习的发展为材料研发开辟了新路径,正逐步实现材料性能的高效精准预测。尤其是机器学习势方法,通过学习第一性原理计算数据集,能够构建兼具高准确性与高效率的势能模型,为大尺度材料动力学模拟提供了可能。
针对当前国内外材料数据库中物理张量数据较为稀缺的问题,唐老师基于图神经网络和高通量第一性原理计算,开发了专门的介电和压电张量数据库平台,并搭建了回归机器学习模型自动化框架,成功应用于光电和热电性能预测。为进一步降低神经进化势能方法的应用门槛,唐老师开发了可视化数据集管理工具NepTrainKit。该工具采用创新的卡片组合设计,极大提升了数据处理效率。这些工具为推动人工智能在材料科学领域的深入应用提供了新的技术支持。
固态电解质失效机制与改性策略的多尺度模型研究

唐法威 科学指南针-唯理计算首席工程师
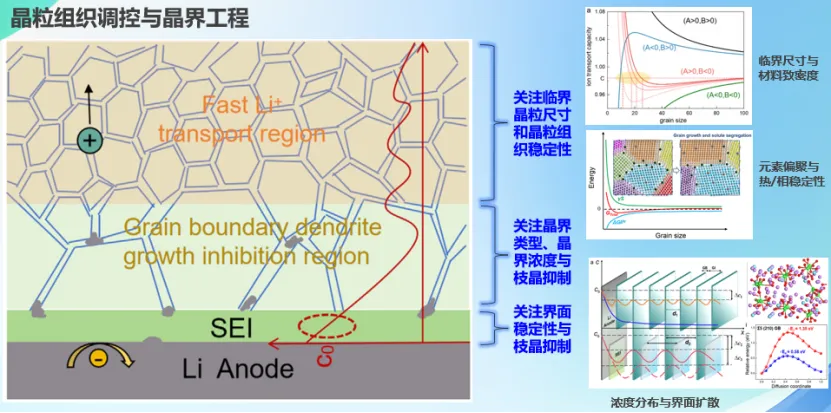
随着电动汽车和储能技术的飞速发展,高能量密度、高安全性的固态锂电池成为下一代电池的焦点。固态电解质凭借高离子电导、宽电化学窗口和强力学性能备受关注。然而,充放电过程中易发生枝晶生长使其实际应用受限于循环寿命较短等问题。
基于此,唐老师围绕固态电解质离子传输动力学和枝晶形核机制,构建了多尺度模型。引入晶粒尺寸效应定量描述了多晶固态电解质中离子电导率随晶粒尺寸的变化关系,揭示了多因素耦合下晶界对离子电导率的影响机制。结合第一性原理计算和数理模型,从微观尺度探讨了晶界对锂传输特征的影响及枝晶形核的作用机制。进一步地,通过有限元模拟从宏观尺度论证了特殊锂传输形式存在的可能性,进而提出多晶体固态电解质材料晶粒组织的非对称设计策略,为开发兼顾高离子电导率和枝晶抑制能力的固态电解质提供了理论指导。
从算法革新到机器学习,从大尺度模拟突破到人工智能与材料设计的深度融合,12 位专家的深度分享和思想碰撞,获得了现场参会者的高度认可。未来,科学指南针将持续深耕前沿科技的探索和研究,持续搭建更高质量的学术交流平台,我们也期待能与全球科研工作者一起,将交流碰撞中迸发的智慧火花转化为技术突破的动力,为科学进步与产业创新注入更持久的能量。








