









 您已经拒绝加入团体
您已经拒绝加入团体

 2026-07-01
2026-07-01
 15
15
 0
0
【摘要】 本文由科学指南针唯理计算围绕 Singleton 2016 年 JACS 经典文献展开解读,解析甲苯硝化区域选择性为何不能由传统过渡态能量或 σ-络合物稳定性解释。研究表明,NO₂⁺ 在显式 CH₂Cl₂ 溶剂和 BF₄⁻ 反离子参与下会经历约 3 ps 的环上游荡,最终选择性由溶剂重排、反离子效应和分子动力学轨迹共同决定,为有机反应机理计算中的模型选择提供参考。

一句话概括:Singleton 用显式溶剂中的分子动力学轨迹,精确复现了甲苯硝化的区域选择性——但这一切和过渡态能量 没有半毛钱关系。NO₂⁺不是从"最优路径"进攻,而是在苯环上空"游荡"了 3 皮秒,最后关头才决定打在哪里。
本文解读的 Singleton 2016 年 JACS 研究表明,甲苯硝化的区域选择性并不能由传统过渡态能量或 σ-络合物稳定性直接决定。通过包含 NO₂⁺BF₄⁻、甲苯和 101 个 CH₂Cl₂ 分子的显式溶剂分子动力学模型,研究发现 NO₂⁺ 在苯环上方会经历约 3 ps 的动态游荡,并在溶剂与反离子重排过程中最终决定进攻位点。完整轨迹模型能够较好复现实验中的邻位、对位和间位比例,而隐式溶剂或忽略反离子的简化模型会显著扭曲选择性。科学指南针唯理计算基于该经典文献,解析显式溶剂分子动力学、反离子效应和反应选择性之间的关系,为有机反应机理计算中的模型选择与动力学分析提供参考。
如果你学过基础有机化学,下面这个结论你一定背过:
甲苯硝化主要得到邻位(~57%)和对位(~41%)产物,间位只有约 2%。教科书解释:邻对位定位效应,σ-络合物更稳定。
这个解释看起来无懈可击。问题是——它根本不对。
不是"不太准",而是物理上站不住脚。Singleton 这篇 JACS 用了一个完整物理模型告诉我们:理解这个反应的选择性,需要的不是过渡态,而是动力学。
一、"薄机制"的破产:为什么过渡态解释是死路
化学家讨论反应机理时,习惯画一条反应坐标曲线:反应物 → 过渡态 → 中间体 → 产物。哪个路径的过渡态能量最低,产物就最多。这叫做"薄机制"(thin mechanism)——只看几个关键静态结构。
但在甲苯硝化这个反应里,薄机制遭遇了全方位的失败:
❌ 标准 DFT 预测翻车现场:
• 用 M06-2X/6-311G*/PCM 计算,预言的 σ-络合物比例为 21% ipso, 20% ortho, 11% meta, 48% para
• 实际实验值:对位只有 41%,而邻位是 57%
• 对位被严重高估,间位被高估了 5 倍
• 就算加上 BF₄⁻ 反离子,预测更糟糕:78% ipso, 19% ortho, 0.1% meta, 3% para ——完全歪了
注意,这里用的不是低水平方法。M06-2X 对这类体系的相对能量偏差只有 0.3 kcal/mol,和 CCSD(T)/aug-cc-pVDZ 对比能量误差仅 0.1 kcal/mol。能量面本身是准的,但预测还是全错。
Singleton 在文中坦率地说:"Even after allowing for trajectory outcomes on a multifurcating surface, the toluene/NO₂⁺/implicit solvent physical model is simply inadequate for predicting or understanding the selectivity."
翻译成中文就是:这条路的尽头是墙。再算过渡态也没用。
二、从静态到动态:让反应自己"演"出来
既然静态结构解释不了,Singleton 换了一个策略。
他搭建了一个 显式溶剂模型:甲苯 + NO₂⁺BF₄⁻ + 101 个 CH₂Cl₂ 分子,直径 25.8 Å。计算方法用了 ONIOM(QM/QM 分层):
计算策略:• 甲苯 + NO₂⁺ → M06-2X/6-311G*(高精度 QM 层)• BF₄⁻ + 101 个 CH₂Cl₂ → PM3(半经验层,负责溶剂和反离子动力学)• 跑 627 条分子动力学轨迹,每条追踪 NO₂⁺ 从远距离接近到最终成键的全过程
先看一个关键结果:
自由能面上根本没有势垒
Singleton 用伞形采样(umbrella sampling)做了 1 ns 的 MD 和 40 万步的 Monte Carlo 来算 PMF(平均力势)。结论令人震惊:
❗ 在 C–N 距离 2.0–4.5 Å 范围内,不存在任何自由能势垒。
NO₂⁺ 一旦进入 4.5 Å 以内,形成 σ-络合物就是不可逆的下坡路——从热力学角度看,反应应该瞬间完成。
但实际轨迹里,它需要3PS(3000 fs)才走完这段"下坡路"。慢了一个数量级以上。3 皮秒听起来很短,但对一个"无势垒"的离子-分子反应来说,这太慢了。正常预期是百飞秒级别。
三、NO₂⁺ 的"环上游荡":选择性是最后一刻才定下来的
轨迹里真正有趣的是 NO₂⁺ 的运动方式。
它不是直奔某个碳原子成键,而是在苯环平面上方游荡:
NO₂⁺ 的"漫游"行为(627 条轨迹统计):
• 每条轨迹中,距离最近的碳原子平均切换27 次
• 最后一次"选边"发生在成键前260 fs,此时 C–N 距离中位数2.42 Å
• 85%的轨迹中,NO₂⁺ 在最终选择前已经逼近到另一个碳原子 2.3 Å 以内
• 25%的轨迹中,NO₂⁺ 曾逼近到某个"最终没有反应"的碳原子2.0 Å以内
• 每当 NO₂⁺ 逼近到某个碳 2.0 Å,有50% 的几率会退回去,而不是直接成键
核心画面:NO₂⁺ 像在环上"挑位置"——反复接近、试探、退回,直到溶剂和反离子 BF₄⁻ 的重排恰好创造了合适的条件。
这不是一个"哪个过渡态能量更低"的故事。这是一个"溶剂重排需要时间,而选择性在等待中被决定了"的故事。
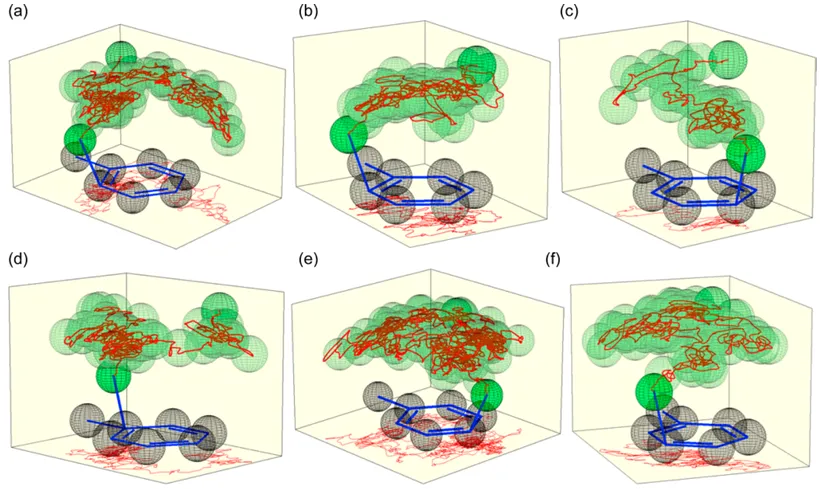
NO₂⁺ 氮原子在甲苯环上方的 3D 运动轨迹
四、轨迹预言 vs. 实验:精准命中
那么这 627 条轨迹算出来的结果到底准不准?直接看表:

* Ipso 加合物会自动 1,2-迁移重排为邻位产物,所以实验邻位 57% 实际 = ortho + ipso。
** 在乙酸酐中捕获 ipso 加合物的实验值。
*** 轨迹预测的 ortho + ipso = 53%,靠近实验的 57%。
✅ 除邻位差约 4 个百分点外,轨迹预测在所有位置上均与实验一致。
反观 σ-络合物平衡分布(上表最后一行),对位高达 98%,邻位只有 2%——这个"热力学控制"的预测和实验完全南辕北辙。
结论:硝化反应的选择性是动力学控制的,但不是过渡态动力学——是溶剂重排动力学。
五、撕掉溶剂和反离子,选择性就崩了
Singleton 还做了一个"控制实验"——把模型简化掉看看结果如何:
|
模型 |
间位 meta |
本位 ipso |
成键时间中位数 |
|---|---|---|---|
|
完整模型 (显式 CH₂Cl₂ + BF₄⁻) |
2.2% |
3.4% |
~3000 fs |
|
模型 A:PCM 隐式溶剂 + BF₄⁻ |
16% |
12% |
~500 fs |
|
模型 B:显式 CH₂Cl₂、无 BF₄⁻ |
18% |
4% |
~400 fs |
|
模型 C:PCM 隐式溶剂、无 BF₄⁻ |
23% |
25% |
~300 fs |
规律非常清楚:
简化越多 → 选择性越差 → 反应越快。
• 少了显式溶剂,间位从 2% 飙到 16–23%
• 少了 BF₄⁻,ipso 从 3.4% 飙到 25%
• 少了这两者,成键时间从 3 ps 缩到 300 fs(10 倍差距)
这解释了为什么常规 DFT 算不准这个反应:你缺的不是更好的泛函,而是溶剂和反离子的真实动力学。
%20PCM%20%E9%9A%90%E5%BC%8F%E6%BA%B6%E5%89%82%20%E2%80%94%20%E9%97%B4%E4%BD%8D%E9%A3%99%E5%88%B0%2016%25(b)%20%E6%98%BE%E5%BC%8F%E6%BA%B6%E5%89%82%E4%BD%86%E6%97%A0%20BF%E2%82%84%E2%81%BB%20%E2%80%94%20ipso%20%E9%A3%99%E5%88%B0%2025%25.jpg)
简化模型下的 NO₂⁺ 轨迹对比:(a) PCM 隐式溶剂 — 间位飙到 16%(b) 显式溶剂但无 BF₄⁻ — ipso 飙到 25%
六、这篇文章对我们做计算的意味着什么?
Singleton 这篇 JACS 2016 不仅是物理有机化学的里程碑,也给做第一性原理计算的人敲了几个很响的警钟:
1. "过渡态能量 → 选择性" 这个逻辑链在离子反应中可能完全失效
你花几天时间找 TS、算 IRC、比能量,最后得出来的选择性和实验不符——可能不是方法问题,是物理模型本身就不对。当反应没有明确的"瓶颈过渡态"、而是在无势垒面上缓慢爬行时,过渡态理论的前提就不成立。
2. 隐式溶剂模型对于离子反应的选择性预测是危险的
PCM/COSMO 等隐式模型能给出合理的反应能垒,但它们在选择性问题上会系统性地出错——因为它们缺少溶剂分子真实重排的时间尺度信息。甲苯硝化只是一个典型案例,类似的"离子 + 芳烃"反应(如傅克反应、卤化)可能面临同样问题。
3. 反离子不只是"电荷平衡"
BF₄⁻ 在轨迹中不是看客。它的位置变化直接决定了 NO₂⁺ 的接近方向和时机。常规计算里把反离子忽略或用隐式溶剂替代,等于丢掉了选择性决定因素的一半。
4. 如果条件有限,至少要知道在算什么
不是每个人都能跑 627 条显式溶剂 QM/MM 轨迹。但读过这篇论文,你至少知道:用隐式溶剂算出来的甲苯硝化选择性大概率是错的,而且错误方向是可预测的——对位会被高估,间位和邻位会被低估。在写论文讨论部分时别硬掰"我们的计算支持实验",而是老老实实说明模型的局限。
文献信息
Nieves-Quinones, Y.; Singleton, D. A. J. Am. Chem. Soc.2016, 138, 15167–15176.
DOI: 10.1021/jacs.6b07328
FAQ
1.甲苯硝化的区域选择性为什么不能只看过渡态能量?
传统过渡态理论通常认为,哪条反应路径的过渡态能量更低,哪类产物就更多。但 Singleton 的 JACS 研究表明,在甲苯硝化中,NO₂⁺ 接近芳环后并不存在清晰的自由能势垒,而是在无势垒反应面上经历显式溶剂和反离子参与的动态运动。选择性不是由某个静态过渡态结构单独决定,而是在 NO₂⁺ 靠近、游荡、退回、再接近的轨迹过程中逐步形成。
2.NO₂⁺ 在甲苯硝化中为什么会“游荡”?
在显式 CH₂Cl₂ 溶剂和 BF₄⁻ 反离子存在下,NO₂⁺ 并不是直接冲向某一个碳原子形成 C–N 键,而是在苯环上方反复接近不同位置。轨迹统计显示,NO₂⁺ 最近碳原子会多次切换,最终成键位点往往在反应最后数百飞秒才确定。这说明溶剂重排和反离子位置变化会显著影响 NO₂⁺ 的运动方向和成键时机。
3.显式溶剂和反离子为什么会影响硝化反应选择性?
显式溶剂和反离子不仅影响反应物的稳定性,还会改变 NO₂⁺ 在芳环附近的空间分布、运动时间尺度和接近方向。Singleton 的模型对比显示,当去掉显式溶剂或 BF₄⁻ 反离子后,反应会明显加快,但区域选择性会严重偏离实验结果。这说明在离子型芳香取代反应中,溶剂和反离子不是背景环境,而是选择性形成过程的一部分。
4.这篇 JACS 文献对有机反应计算有什么启示?
这篇文献说明,对于某些离子反应、无势垒反应或多分叉反应面体系,单纯寻找过渡态、比较能量和做 IRC 可能不足以解释实验选择性。更合理的做法是根据体系特征引入显式溶剂、反离子、分子动力学轨迹、自由能采样或 QM/MM 模型,从动态过程而不是单一静态结构理解反应机理。
5.科学指南针唯理计算可以为类似研究提供哪些计算思路?
对于涉及离子反应、溶剂重排、反离子效应和区域选择性的体系,可以考虑结合 DFT、显式溶剂模型、分子动力学轨迹、自由能采样和 QM/MM 分层计算等方法,评估静态能量模型是否足以解释实验结果。科学指南针唯理计算可围绕具体课题需求,协助设计更贴近真实反应环境的模拟方案,用于分析反应路径、选择性来源和溶剂/反离子的动态作用。








