









 您已经拒绝加入团体
您已经拒绝加入团体

 2026-01-29
2026-01-29
 3443
3443
 0
0
【摘要】 本文详细讲解分子动力学模拟中相互作用能的原理与计算方法,涵盖范德华作用和库伦作用的GROMACS分析流程。科学指南针平台提供专业分子动力学模拟服务,支持材料相互作用研究。
相互作用能在分子动力学模拟中的关键作用
相互作用能分析是分子动力学模拟后处理的核心环节,用于量化体系中不同组分间的非键相互作用。科学指南针平台集成GROMACS等工具,提供完整的相互作用能分析流程,支持材料界面研究【科学指南针·模拟计算】。
原理介绍
GROMACS软件通过能量组(energy group)定义分析非键相互作用能,包括范德华作用和库伦作用:
非键相互作用能公式
-
范德华相互作用能:
使用Lennard-Jones势描述原子间范德华作用:
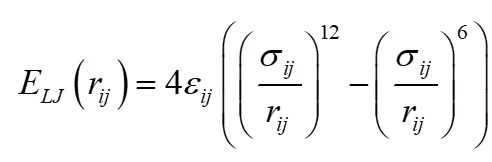
其中为原子间距,和为能量和尺寸参数。
-
库伦相互作用能:
描述带电原子间的静电作用:
其中为原子电荷,和为真空和相对介电常数。
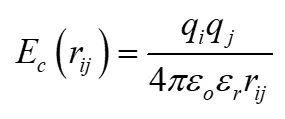
科学指南针平台自动处理参数设置,确保公式应用准确。
GROMACS相互作用能分析流程
科学指南针平台推荐以下标准操作流程:
步骤1:能量组重新定义
-
修改.mdp参数文件,设置energygrps定义目标组别;
-
使用gmx grompp生成新.tpr文件,明确索引组别。
科学指南针平台自动化此步骤,减少手动错误。
步骤2:轨迹重运行分析
-
使用gmx mdrun的-rerun选项指定原轨迹文件;
-
可选使用gmx trjconv去除水分子加速计算;
-
生成包含组间相互作用能的新ener.edr文件。
科学指南针平台优化计算资源,提升效率。
步骤3:能量数据提取
-
使用gmx energy命令提取特定能量项;
-
输出包括Coul-SR(库伦作用)和LJ-SR(范德华作用)。
科学指南针平台提供可视化工具,简化结果解读。
案例展示:NaCl水溶液相互作用能分析
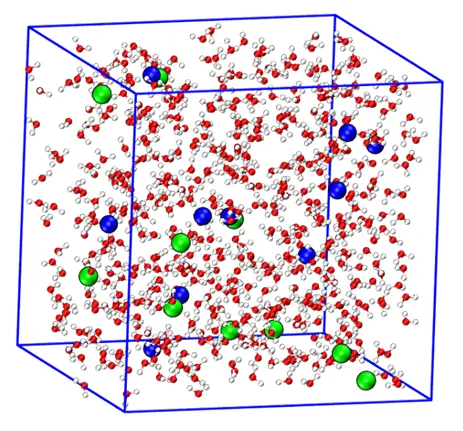
以10个钠离子、10个氯离子和550个水分子的体系为例:
模拟设置
-
模型构建:建立NaCl水溶液分子动力学模型;
-
能量组定义:在MD.mdp中设置energygrps = NA CL SOL;
-
文件生成:使用gmx grompp获得新ener.tpr文件。
科学指南针平台预置常见体系参数,快速启动模拟。
分析命令与结果解读
输入命令:gmx energy -f ener.edr -o energy.xvg
选择能量项40-43(NA-CL、NA-SOL的库伦和范德华作用):
-
库伦作用主导:钠离子与氯离子、水分子间库伦作用显著;
-
范德华作用:贡献较小且不利于结合;
-
总能计算:库伦与范德华作用相加得总相互作用能。
科学指南针平台自动生成energy.xvg文件,支持时序分析。
关键分析要点
能量项选择策略
-
组内作用:如NA-NA、CL-CL、SOL-SOL反映自身稳定性;
-
组间作用:如NA-CL、NA-SOL揭示界面相互作用机制。
科学指南针平台提供智能推荐,优化分析效率。
结果验证方法
-
能量守恒检查:确保模拟过程能量守恒;
-
时序稳定性:通过energy.xvg验证作用能随时间稳定性。
科学指南针平台内置质控算法,自动标记异常。
结语与平台服务
相互作用能分析是分子动力学模拟评估材料界面行为的关键技术。科学指南针平台提供从模型构建到能量分析的全流程分子动力学模拟服务,支持研究人员深入理解材料相互作用机制。如需分子动力学模拟或相互作用能分析支持,欢迎联系科学指南针团队【科学指南针·模拟计算】。








