









 您已经拒绝加入团体
您已经拒绝加入团体

 2025-12-02
2025-12-02
 3124
3124
 0
0
【摘要】 本文汇总Nature和Science最新电池研究成果,涵盖孙学良团队锂离子导体、大连化物所氢负离子电池、西湖大学SEI机理突破,并详解电池热力学、动力学、正极材料、电解液计算四大专题,附孙成华教授电池计算课程信息。

最近电池材料成果频繁登上Nature\Science正刊,以下是3篇重要研究成果:
(一)三院院士孙学良团队,再发Science
加拿大西安大略大学(东方理工学院)孙学良院士团队联合美国马里兰大学莫一非教授等研究者通过混合阴离子设计策略,开发了结晶Li3Ta3O4Cl10(LTOC)及其衍生物,这些物质具有优异的离子电导率(在25°C时高达13.7毫西门子每厘米)和电化学稳定性。
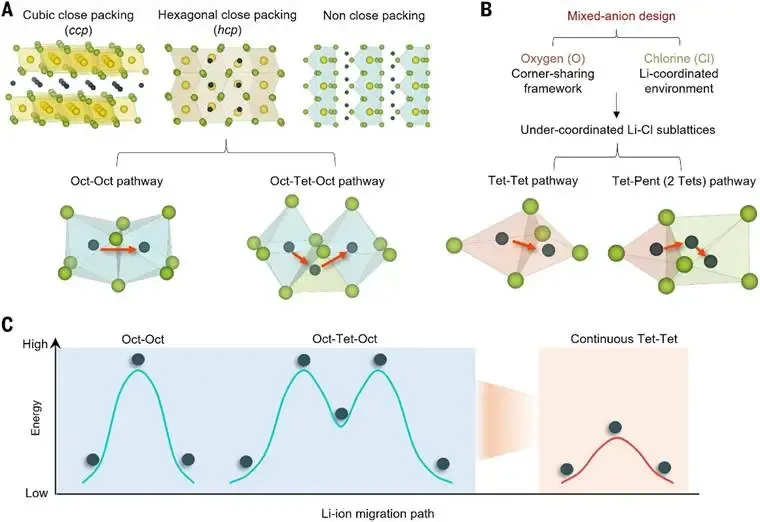
图 阴离子亚晶格设计朝向低能垒锂离子迁移路径的示意图 © 2025 AAAS
(二)仅3张图,中科院大连化物所,重磅Nature
中国科学院大连化学物理研究所的张炜进、曹湖军、陈萍等研究者开发了一种核壳结构的氢化物材料 3CeH3@BaH2,其在室温下表现出快速的氢负离子(H-)传导性能,并在 60 °C 以上转变为超离子导体。使用氢作为电荷载体可避免有害的金属枝晶形成,这为清洁能源存储与转换开辟了新的研究路径。
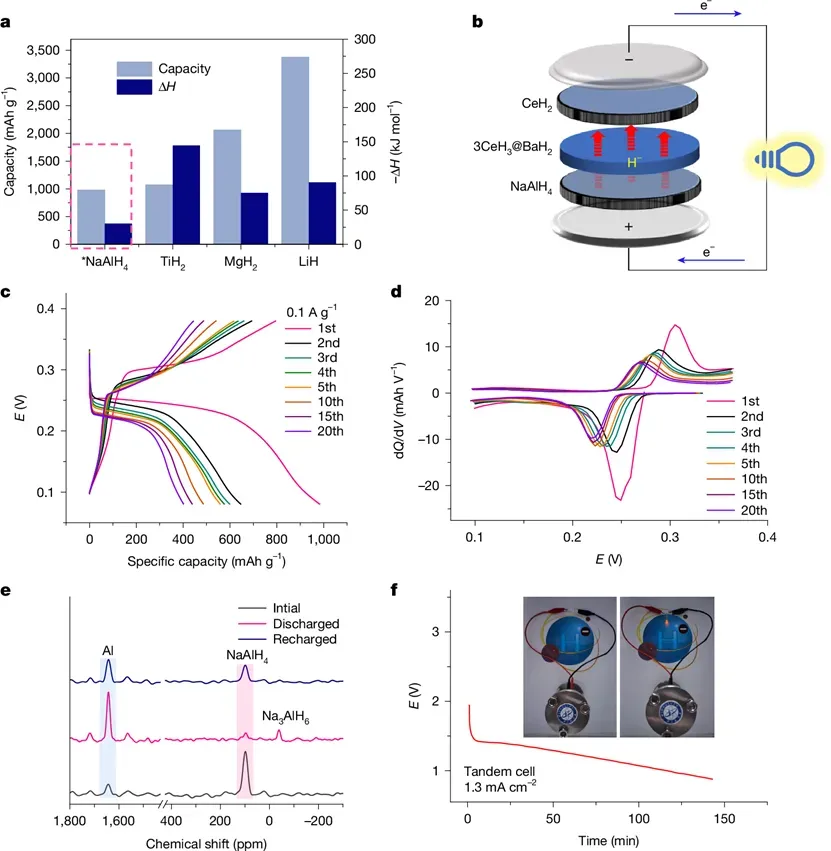
图 全固态可充电氢负离子电池 CeH2 | 3CeH3@BaH2 | NaAlH4 的电化学性能 © 2025 Springer Nature Limited
(三)西湖大学&复旦大学 最新Nature
西湖大学工学院向宇轩课题组、朱一舟课题组及复旦大学宋云等研究者首次通过19F固态核磁共振(NMR)、6Li同位素NMR等技术证实,SEI中的LiF并非传统认知中的纯相,而是形成了包含富氢相(LiH1-yFy)和富氟相(LiF1-xHx)的有限LiF-LiH固溶体,同步辐射X射线衍射、冷冻电子显微镜进一步验证了该固溶体的相分离特征,打破了“SEI中LiF为单一纯相”的传统认知。
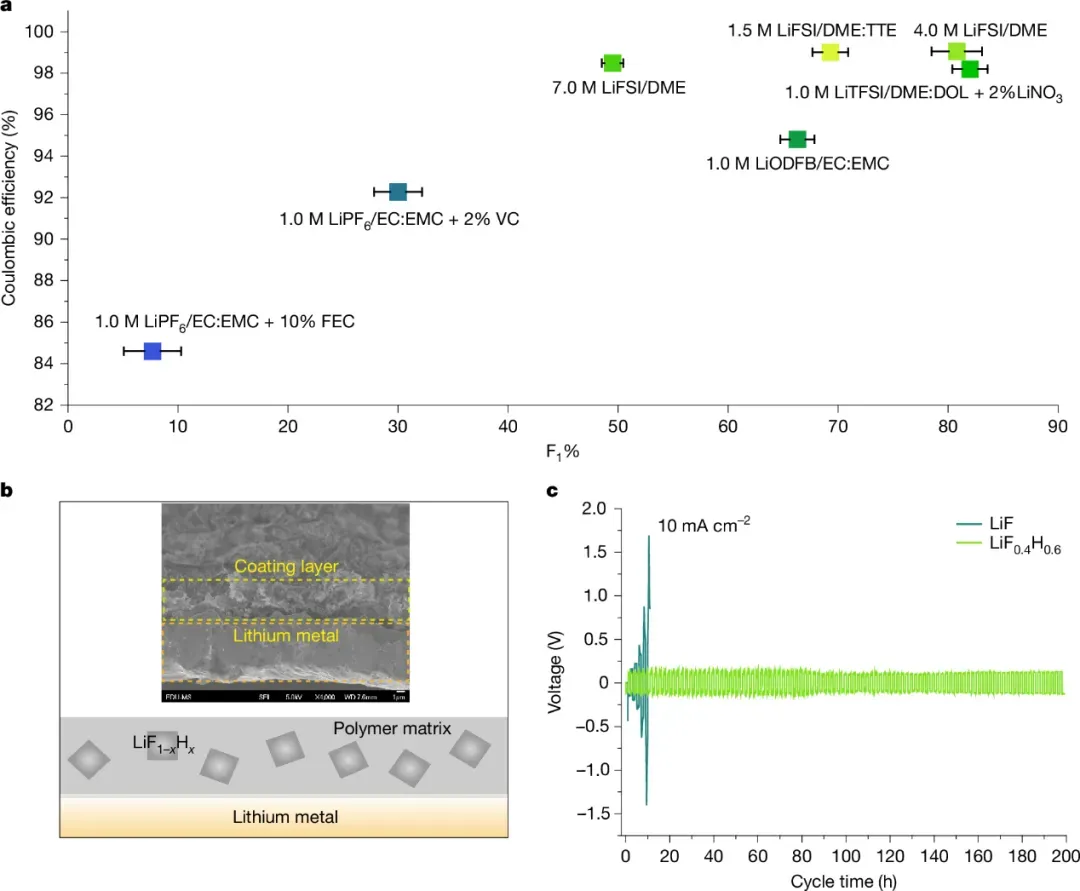
图 超越富LiF固体电解质界面设计 © 2025 Springer Nature Limited
专题一 电池热力学
电极热力学原理与计算框架
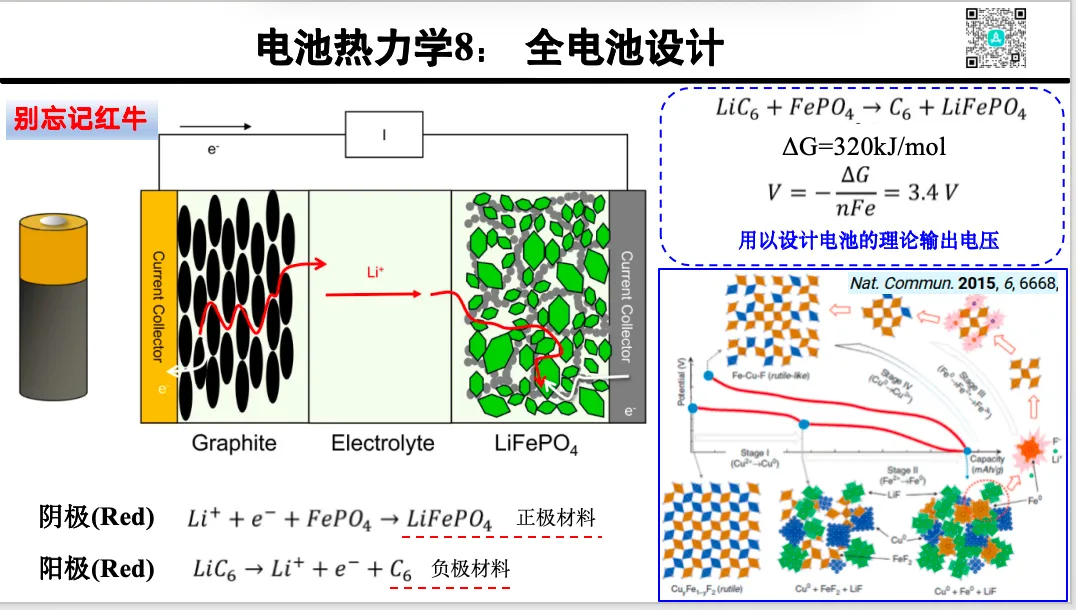
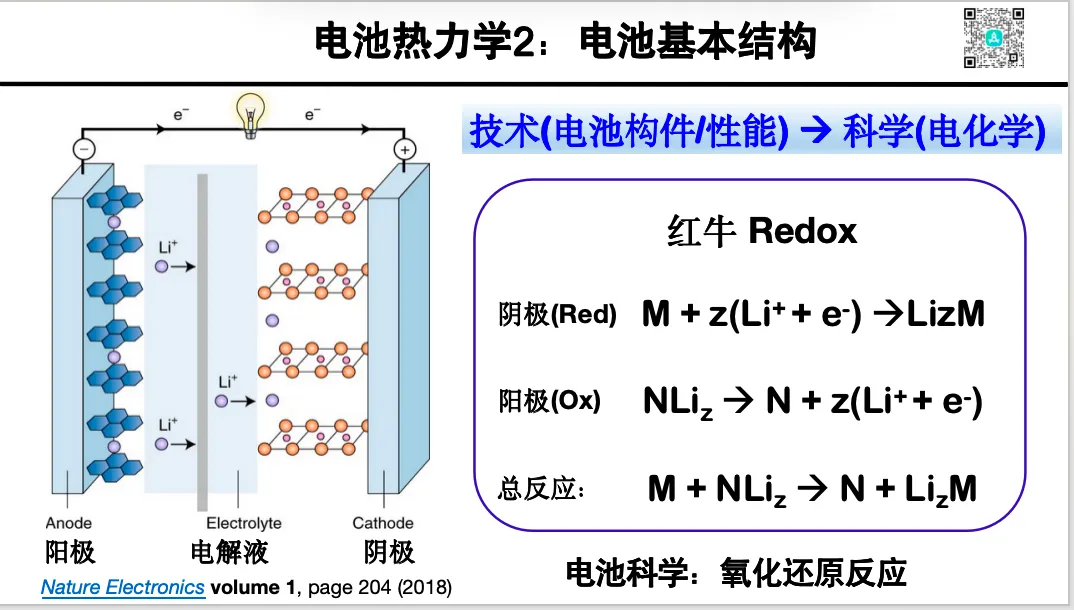
1. 电极反应的热力学基础:自由能 化学势与电压关系
2. 电池中的第一性原理计算热力学的切入点
3. 电极稳定性判据:形成焓、分解电压、相稳定性图
4. 缺陷热力学与掺杂稳定性分析
5. 从能带结构与态密度解读电极热力学行为
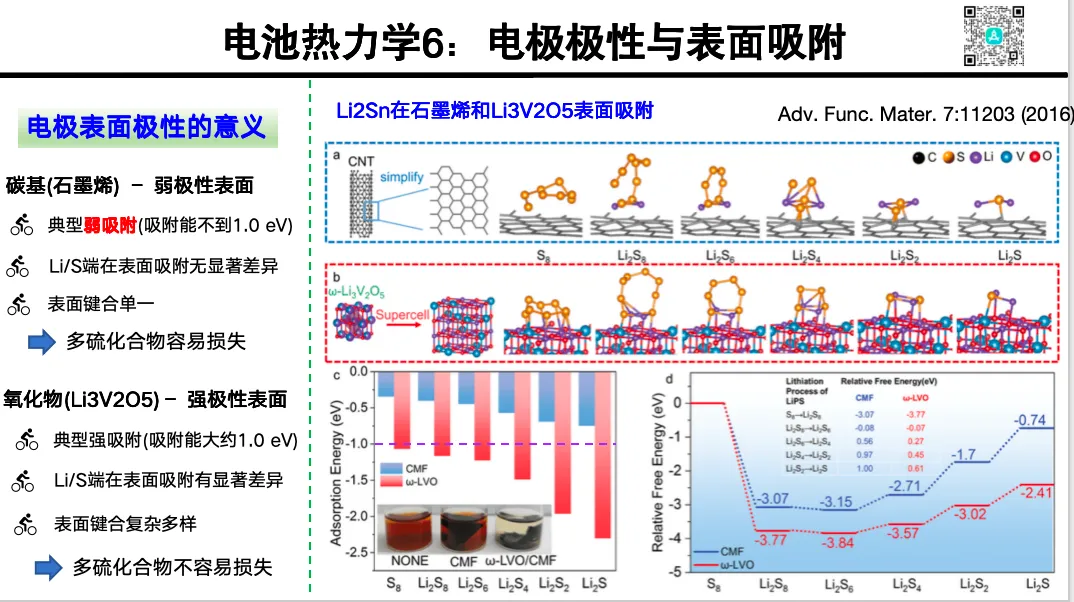
电极表面吸附热力学
1. 单金属离子吸附热力学
2. 溶剂分子的吸附能与构型
3. 多硫化物的吸附
4. 锂离子 / 溶剂共吸附
5. 带电离子吸附的能量误差问题与校正思路
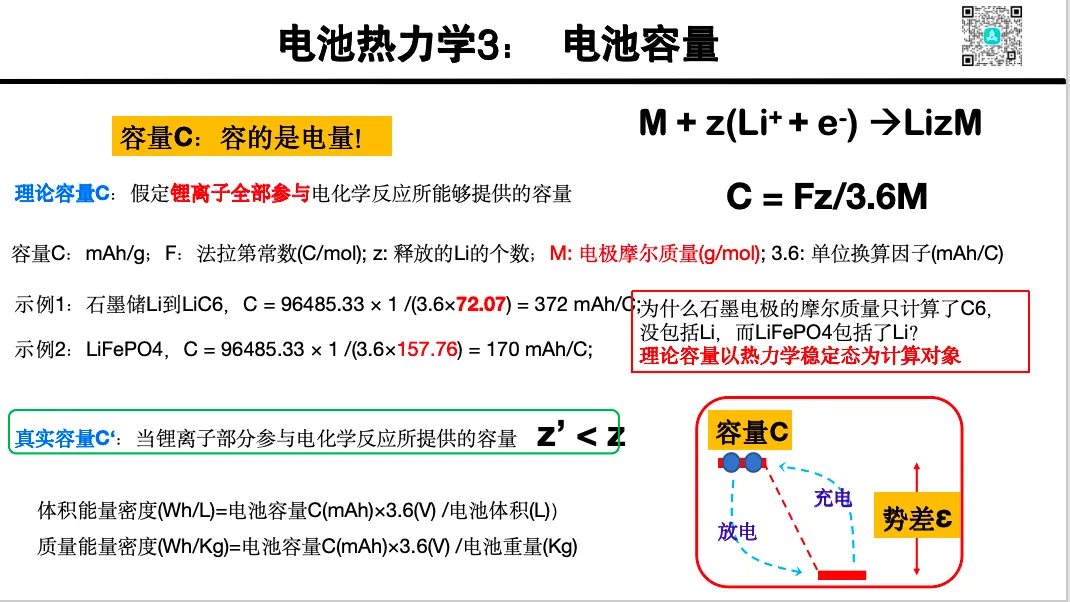
充放电过程热力学
1. 充放电的氧化还原机制与能量公式推导
2. 离子嵌入构型扫描与电压曲线预测
3. 容量计算与结构变化的热力学解释
4. 过充、结构膨胀与失稳
5. 电压平台与充放电时的相变问题
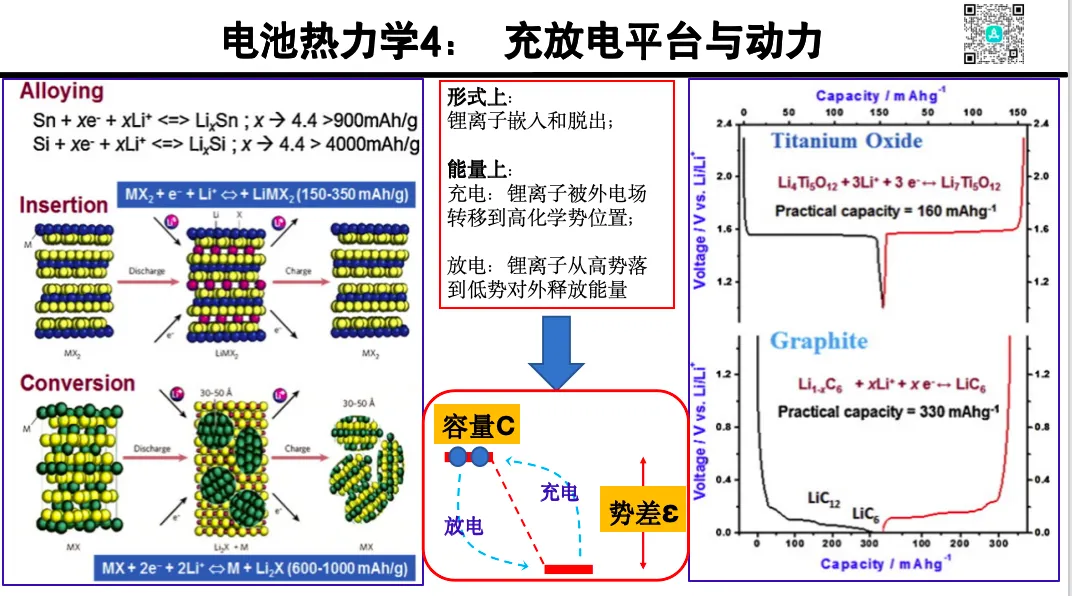
电池系统热力学前沿方法
1. DFT 计算结构和能量时的三个关键指标
2. DFT+U 与 SCAN 泛函对电极热力学的校正应用
3. 第一原理分子动力学(AIMD)分析热稳定性
4. 热力学与动力学耦合:从稳定性到速率
5. 材料数据库对电池设计中的应用
操作课:电极与表面热力学计算(经典正极电池热力学)
1. 典型正极(LiCoO₂)结构优化与电子结构分析
2. 形成焓计算与热稳定性判据
3. DFT+U 参数调优对电子结构分析的影响
4. 正极过充 / 过放情形下的失稳机制分析
5. 电极失稳分解热力学快速计算
操作课:充放电热力学模拟(经典负极表面热力学)
1. 锂离子在石墨电极表面的吸附能(带电离子如何处理)
2. Li₂S 的单 / 多分子吸附
3. 构建不同嵌锂程度的石墨负极模型(Li⁺分布隐含的作用类型)
4. 计算充放电曲线与容量变化
5. 负极过充条件下的热力学失稳分析
上机实操巩固 上机操作、自由交流、群内答疑
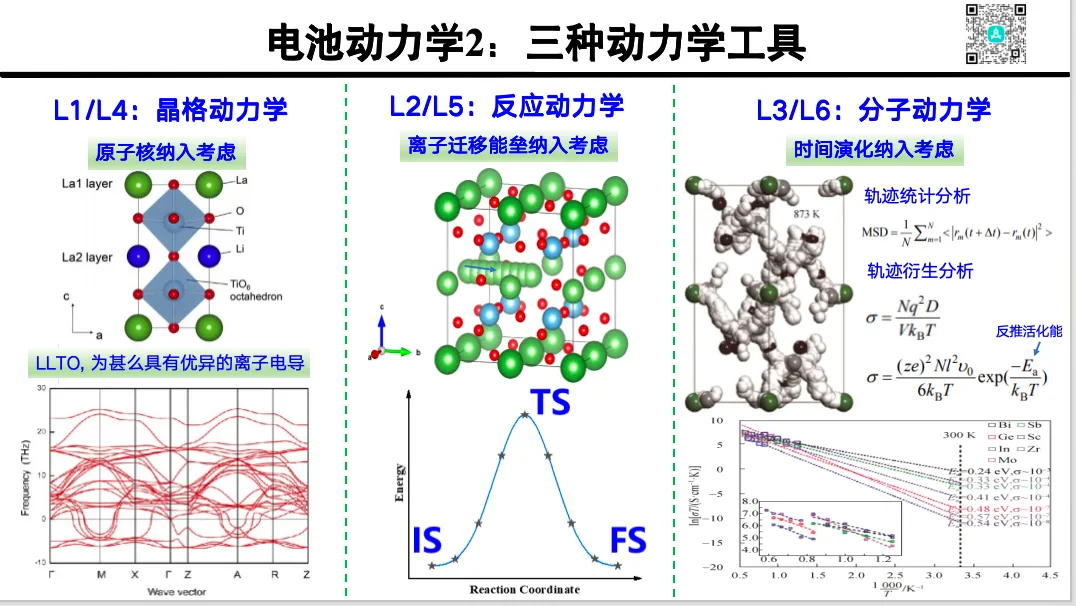
专题二 电池动力学
电极动力学的物理基础与衡量参数
1. 电极稳定性判据理论
2. 电池动力学理论模型(Butler–Volmer, Marcus–Hush 理论)
3. 固态界面相(SEI/CEI)的形成与演化动力学
4. 电荷转移步骤的速率常数与过电位关系
5. 马库斯理论与电荷转移
电极内部扩散动力学
1. 固相扩散系数定义与测算方法(Fick定律,Nernst–Planck方程)
2. 锂离子在晶格中的迁移路径与能垒
3. 缺陷对离子扩散动力学的调控
4. 多粒径与多相结构中的扩散非均匀性
5. 非晶电极中的锂离子扩散特殊性
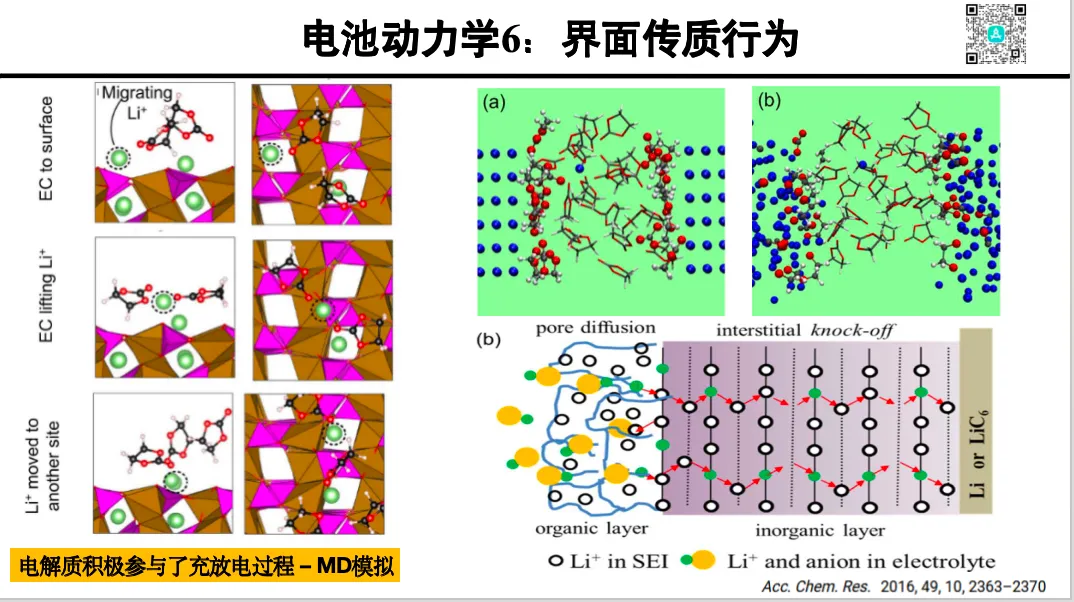
电极界面动力学
1. 界面吸附–脱附平衡
2. 界面电场对反应速率的影响
3. 溶剂化对离子迁移动力学的影响(第一原理分子动力学)
4. 电极极化对界面反应的调节
5. 构建反应路径图(Reaction Pathway Map)的方法
电极晶格动力学
1. 电极稳定性与晶格动力学
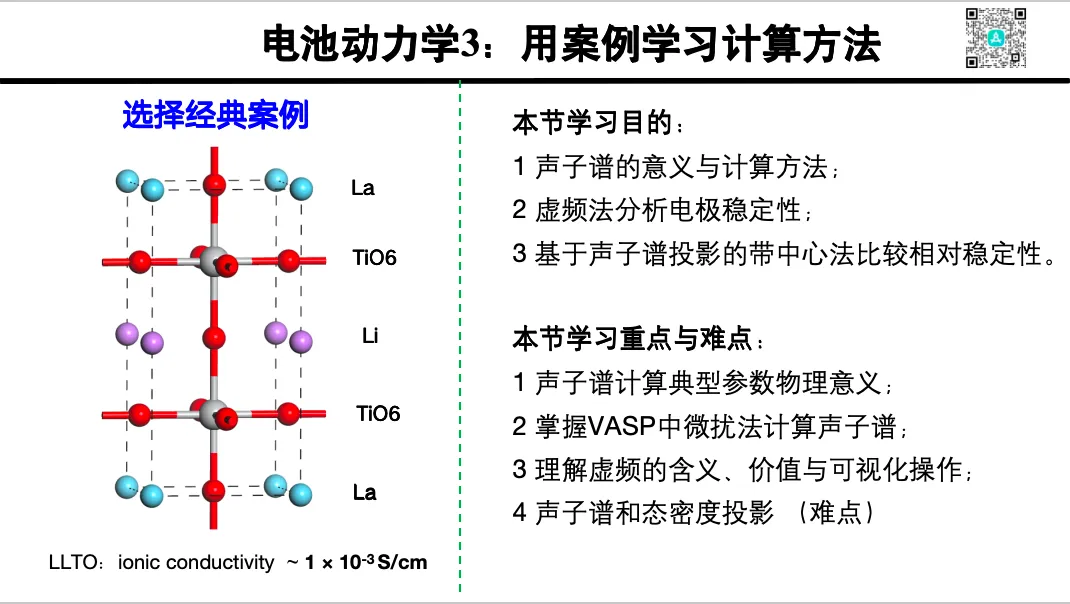
2. 声子与声子谱计算
3. 声子谱中心法判断稳定性
4. 声子谱与变温自由能
5. 声子谱中的虚频分析
操作课:从第一性原理到动力学参数提取
1. AIMD 基础与在 VASP 中的参数设计
2. 锂离子在石墨烯表面的室温迁移(表面动力学)
3. 锂离子在 LLTO 电极中的块体迁移(块体动力学)
4. 扩散系数的 AIMD 计算与拟合
5. 离子扩散路径对比与阻力机理分析
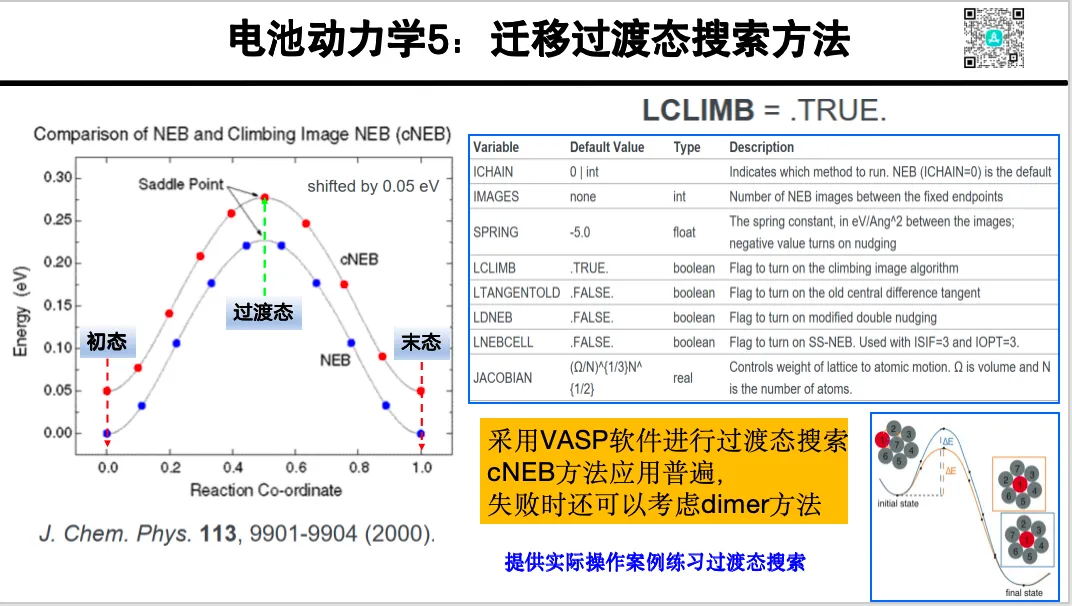
操作课:微观机理反应力学 - 过渡态与 NEB 搜索
1. 过渡态理论与搜索方法
2. NEB 参数和中间态插点技巧
3. 锂离子在石墨烯表面迁移动力学
4. 阳离子空位下的锂迁移
5. 迁移路径的可视化
上机实操巩固 上机操作、自由交流、群内答疑
专题三 锂电池正极材料计算与分析
经典层状正极
1. 锂电池正极概览
2. 经典层状 LiCoO₂和 LiNiO₂正极
3. 三元锂正极
4. 阳离子混排问题
5. 正极中的 d-p 杂化问题
尖晶石与橄榄石型正极
1. 尖晶石正极(LiMn₂O₄)
2. 橄榄石正极(LiFePO₄)
3. 阳离子的姜泰勒效应与稳定性问题
4. 阳离子 redox 电压平台和电荷补偿效应
5. 阴离子化学
高镍、富锂、聚阴离子正极
1. 高镍氧化物正极
2. 富锂层状氧化物(Li₁₊ₓM₁₋ₓO₂)正极
3. 晶格氧稳定性与 O₂释放
4. 岩盐 - 尖晶石相转变问题
5. NASICON 正极材料
高电压、多电子与多氧化还原中心正极
1. 正极如何提升工作电压
2. 多电子反应正极
3. 多氧化还原对之间的竞争关系
4. 硫化物、氟化物正极
5. 化学键的离子性和共价性"
操作课:层状与尖晶石正极材料的计算流程(经典体系)
1. 构建 LiMn₂O₄晶体结构模型
2. 计算晶格常数与体积变化
3. 电压曲线计算与结果可视化
4. DOS、PDOS 分析与电子态变化解释
5. 氧空位形成能与结构稳定性分析
操作课:高镍与前沿正极体系的模拟(前沿体系)
1. 构建高镍 NCM 与富锂层状氧化物模型
2. 计算 Ni 价态变化与基于 Bader 电荷的定量评估
3. 晶格氧稳定性(电子结构评估)与氧空位形成能(能量评估)
4. 多 Redox 中心时的电子结构与竞争示意图
5. 基于配位场的 d-p 杂化分析
上机实操巩固 上机操作、自由交流、群内答疑
专题四 电解液计算与分析
电解液分子与离子的结构与性质计算
1. 电解液在电池中的功能与重要性
2. 常见电解液分子(酯类、醚类、离子液体)及溶剂化离子
3. 电解液分子几何结构与电荷分布
4. 分子轨道与 HOMO–LUMO 间隙在电化学稳定性预测中的作用
5. 溶剂化能、偶极矩与极化率计算
电解液 / 电极界面吸附与电子结构
1. 电解液分子在电极表面的吸附构型与能量计算方法
2. 电荷转移分析(Bader、差分电荷密度)
3. 电极表面局域电子态密度(PDOS)对分子稳定性的影响
4. 电场作用下界面电子结构变化
5. 从吸附能与电子态预测界面副反应趋势
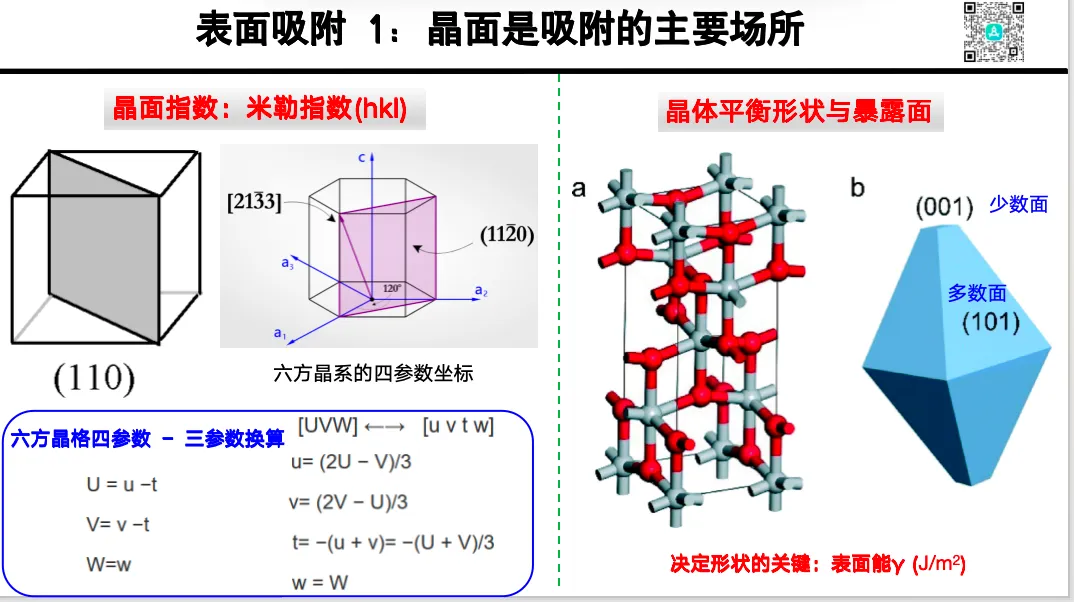
电解液 / 锂盐微观作用与离子迁移
1. 典型锂盐结构和基础参数
2. 锂盐单体在电解液块体中的溶剂化模拟
3. 锂盐 / 电解液在电极表面共吸附模拟
4. 第一性原理模拟锂离子在电解液中的扩散
5. 过渡态搜索锂离子在电解液 / 电极界面迁移行为
电解液的电化学稳定性与分解机理模拟
1. 电化学稳定窗口的定义与计算原理
2. 氧化 / 还原反应能垒与反应路径(CI-NEB、TS 搜索)
3. 分子动力学(AIMD)模拟电解液分解过程
4. 温度、压力与溶剂环境对分解反应的影响
5. 电解液添加剂在分解抑制中的作用机理

操作课:电解液分子与电极界面计算
1. 构建并优化常见电解液分子与溶剂化离子模型
2. 计算分子电荷分布与 HOMO–LUMO 能隙
3. 构建电解液分子在电极表面的吸附构型
4. 计算吸附能与差分电荷密度
5. 分析吸附前后 PDOS 变化
操作课:电解液界面稳定性与分解反应模拟
1. 构建电解液 / 固态电解质界面模型
2. 优化界面结构并计算结合能
3. 使用 NEB 计算离子迁移能垒
4. 基于 AIMD 模拟锂离子扩散系数
5. 电解液的界面分解反应计算
上机实操巩固 上机操作、自由交流、群内答疑
讲师介绍
孙成华教授,毕业于中国科学院,曾在哈佛大学、普林斯顿大学从事博士后研究。从事计算模拟25年,已发表400余篇SCI论文,论文引用超过3万次,高被引学者,开发40余门计算模拟课程,一对一辅导1000+科研工作者。
适合人群
零基础/初学者:物理、化学、化工、材料、能源、环境等相关专业的本科生和研究生,希望系统学习电池计算方法
有一定基础的科研人:已具备DFT 或 VASP 基础,或从事电池材料模拟与设计的青年教师和科研人员,想深入理解电池反应机制与材料性能,并提升科研产出
课程时间
2025年12月6日-12月9日 (09:00-11:50--14:00-18:00)
线下学习:武汉线下培训
线上学习:全程直播参与
线上和线下区别:
线下:小班学习氛围强,真正沉浸式训练,并且讲师面对面1v1指导,可能之前卡了你几个月的问题,现场十分钟就讲明白了。适合科研卡点多,希望快速突破计算难题的同学。
线上:对于无法出差或时间安排有限的科研人,我们新增了线上直播。提供200页教材,全程参与直播,同时有无限次回放+课后180天答疑权益。

课程费用
报名倒计时1周,目前线上/线下报名还剩少量名额,优惠价格即将截止!
原价4499元 早鸟特惠3999元
上课方式可选线上/线下参与
永久无限次复听权益

扫码咨询
免费试听|免费机时|专属干货
如果你因为时间安排无法到现场,本期课程支持线上参与,同时提供PDF讲义、全套直播课程回放、线上连麦提问、课后答疑等权益。
报名费用可开具正规报销发票及提供相关缴费证明、邀请函等文件用于报销。
增值优惠
组合优惠:符合以下任一情况可在早鸟特惠基础上,再享95折
·每位学员购买2门及以上计算线下课
·2人及以上学员组团报名计算线下课
售后答疑:课后180天内免费社群答疑
机时体验:免费送5000核时机时,多种GPU资源可选
专属赠送:报名即送《模拟计算指南》一本








