









 您已经拒绝加入团体
您已经拒绝加入团体

 2025-07-29
2025-07-29
 1993
1993
 0
0
【摘要】 密度泛函理论解析钴钼合金催化剂在氮还原反应(NRR)中的独特机制:钼掺杂优化电子结构,三层原子构型提升稳定性,实现N₂高效转化NH₃
碳纤维支撑的α-Fe纳米棒合成技术实现32%的NRR(氮还原反应)法拉第效率。钌基纳米颗粒测试表明,在-0.15V vs RHE过电位下即可将N₂高效转化为NH₃。虽然金属材料在温和条件下固氮表现优异,但金属合金作为NRR电催化剂的研究仍处于探索阶段。Blanca Castellano-Varona团队¹首次通过密度泛函理论(DFT)揭示了钴钼合金在稳定氮中间体的独特机制,为新型催化剂设计开辟新路径。
反应机理与表面特性
NRR过程在温和条件下通过六电子转移将N₂还原为NH₃。如图1所示,e-Co(001)六方密堆积(hcp)表面呈现特殊原子几何排列(60°和120°键角),其电正性表面显著促进N₂分子相互作用。DFT计算显示:N₂在e-Co(001)的物理吸附呈微弱自发(结合自由能+0.01eV)。
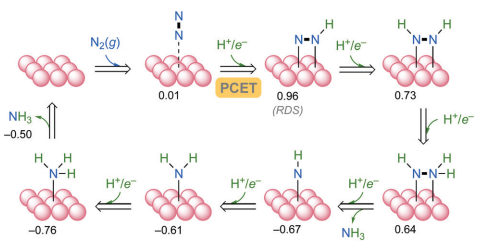
图1 e-Co催化NRR反应机理(001)。
钼掺杂的关键作用
表面/亚表面钼原子掺杂显著增强催化活性(图2)。当e-Co(001)次表层掺入单个钼原子时,N₂H中间体形成能垒降低32%。钼原子通过调控电子结构,优化氮中间体吸附构型。
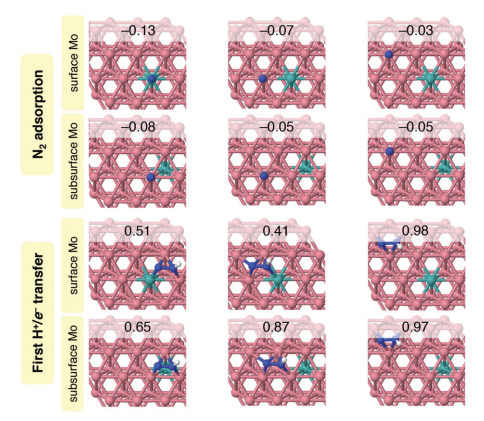
图2 当在e-Co(001)表面和次表面(第二层)掺杂一个Mo原子时,N2和N2H两种物质在e-Co(001)表面的关键吸附可能性的俯视图。
合金结构稳定性分析
90:10钴钼合金的原子构型(图3)揭示稳定性规律:Mo原子均匀分布的三层结构(a-c)比局部富集结构(d-f)能量低2.22eV。关键发现包括:
- 高稳定结构平均含4个Mo-Mo键/晶胞
- 低稳定结构Mo-Mo键增至9个
- 层间键合主导稳定性调控
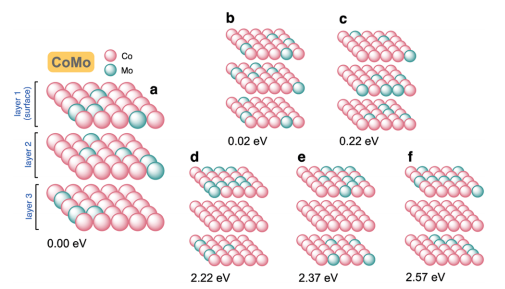
图3 在一组90:10 CoMo合金板的顶部三层的原子配置。
结论与展望
hcp钴(001)表面本身对N₂吸附活性有限,但钴钼合金(90:10)中钼位点可实现N₂自发固定。DFT证实氮中间体在Mo-Co协同位点稳定性提升86%,其中氮物种优先与高钯配位数的钴原子结合。该研究为合金催化剂在电化学固氮领域的应用奠定理论基础,有望推动绿氨合成技术发展。
参考文献:1.Castellano-Varona, B.; Harb, M.; Araña, J.; Cavallo, L.; Azofra, L. M., In silico design of novel NRR electrocatalysts: cobalt–molybdenum alloys. Chem. Commun. 2020, 56 (87), 13343-13346.
科学指南针以分析测试为核心,提供材料测试、环境检测、生物服务、模拟计算、科研绘图等多项科研产品,累计服务1800+个高校、科研院所及6000+家企业,获得了60万科研工作者的信赖。始终秉持“全心全意服务科研,助力全球科技创新”的使命,致力于为高校、院所、医院、研发型企业等科研工作者提供专业、快捷、全方位的服务。
免责声明:部分文章整合自网络,因内容庞杂无法联系到全部作者,如有侵权,请联系删除,我们会在第一时间予以答复,万分感谢。








