









 您已经拒绝加入团体
您已经拒绝加入团体

 2023-12-06
2023-12-06
 4023
4023
 0
0
【摘要】 无参数第一性原理计算,基于密度泛函理论,只需要知道原子种类和晶体结构的知识,即可以进行预测计算。
无参数第一性原理计算,基于密度泛函理论,只需要知道原子种类和晶体结构的知识,即可以进行预测计算。结合拟谐方法,可以用第一性原理计算准确地描述固相的有限温度热力学,其中体积V和温度T所用的Helmholtz自由能F(V,T)通常近似为,
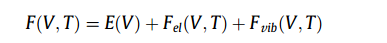
其中E(V)是在0K时的静态能量(无零点振动能)和体积V,通过第一原理计算预测,并由状态方程(EOS)拟合。FEL(V,T)表示相对于相应的V和T的热电子对自由能的作用,这对于金属(而不是半导体和绝缘体)系统尤其重要,因为费米能级上的电子密度不为零。Fvib(V,T)是振动对自由能的贡献,为了精确,通常用声子计算来描述,为了简单和效率,通常用德拜模型来描述。尽管已经有了高压下压力与体积(P-V)状态方程的比较研究,但很少有人注意到不同E-V状态方程的匹配。此外,声子模型和德拜模型的振动贡献的比较研究在文献中也很少。
Shang等人在方程的基础上研究面心立方结构的Ni和L12结构的Ni3Al的第一性原理热力学,并评价了不同状态方程的拟合以及声子模型和德拜模型对振动的作用。他们选择Ni和Ni3Al是由于技术上重要的镍基高温合金,特别是Gleeson和他的同事新开发的涂层比目前的铂修饰NiAl涂层的氧化动力学慢了10-20倍。这些新涂层是以Ni-Al-Pt系中Ni+Ni3Al的两相混合物为基础,再添加以Ni3Al为主相的CrHf、Y和Zr合成的。由于这些新涂层与镍基高温合金具有相同的成分,它们为开发适用于当前和未来一代高温合金的高抗氧化和相容涂层开辟了道路。
免责声明:部分文章整合自网络,因内容庞杂无法联系到全部作者,如有侵权,请联系删除,我们会在第一时间予以答复,万分感谢。








