









 您已经拒绝加入团体
您已经拒绝加入团体

 2023-10-07
2023-10-07
 2665
2665
 0
0
【摘要】 预测固体热性质的能力对于理解光电子和能量转换器件的热管理和设计高效热电材料至关重要。
预测固体热性质的能力对于理解光电子和能量转换器件的热管理和设计高效热电材料至关重要。然而,尚未开发出一种有效且可预测的量子模拟框架来研究固体的热性质,其复杂性与经典模拟相同。针对材料热性能对高效通用量子模拟框架的需求,Marcello Puligheddu等人[1] 提出了一种从第一性原理模拟热传输的方法,该方法可用于复杂、均匀和非均匀固体的预测计算。我们通过实现正弦温度梯度来推广平衡分子动力学(AEMD)方法,从而避免温度不连续性。这种方法称为正弦AEMD。在AEMD方法中,固体的平均温度由不连续的温度分布任意改变;随后通过进行NVE模拟,监测施加扰动后接近平衡的方式。特别地,使用T向平衡值的时间衰减来计算热导率。在计算过程中,使用了局部Nosé-Hooper恒温器来施加初始正弦曲线。
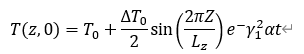
而受正弦T曲线影响的系统两侧的平均温度之差为
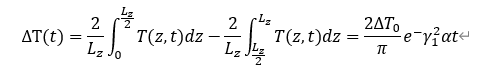
而后,在NVE模拟期间30个MgO晶体样品(1280个原子样品)的瞬时平均温度,以及热方程的解析解(连续线)。数据显示了温度差作为时间的函数和用于计算热扩散率的拟合函数。此外,还进行了单独的计算,以计算热容作为T的函数,导热系数很容易从κ = αρcy中得到,其中p是系统的已知密度(通过选择体积V来设置)。实验数据证明SAEMD可以直接应用于使用密度泛函理论(DFT)计算热导率。这种方法只需要计算MD轨迹和原子力,而不需要额外计算能量导数,例如力常数、能量密度或电流的直接计算。讨论了代表性氧化物MgO在不同温度下以及有序和纳米结构形态下的结果,显示了该方法在不同条件下的性能。
[1] Puligheddu M , Gygi F , Galli G . First-principles simulations of heat transport[J]. Physical Review Materials, 2017, 1(6).
科学指南针通过互联网技术建立更可靠的服务标准,全国30个办事处,19个城市拥有自营实验室,最好的设备、最专业的老师为您服务。








