







 您已经拒绝加入团体
您已经拒绝加入团体

 2021-10-22
2021-10-22
 6056
6056
 2
2
【摘要】 今天我们从CO在Au(111)面上的吸附来说明吸附位点的选择和吸附结构的相关优化
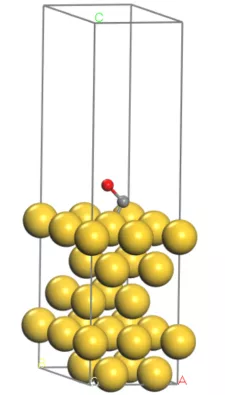
首先我们构建出Au(111)面的结构模型,如下图:
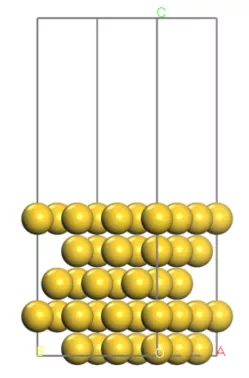
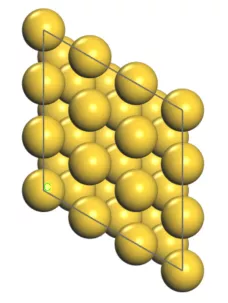
对于Au这种面心立方金属的(111)表面,一般认为存在4种吸附位点:
[1] Top位,就是Au(111)表面原子的上方
[2] Bridge位,两个Top原子的中间,可以理解为在两个Au原子间搭了一座桥。
[3] hollow位,即在三个原子的中心,又可以分为 Fcc位和Hcp位。
具体细化一下就是:Fcc(face centered cubic)指的是表面的Hollow位正下方(即第二层)没有原子,而第三层有原子,则这个hollow为Fcc位。
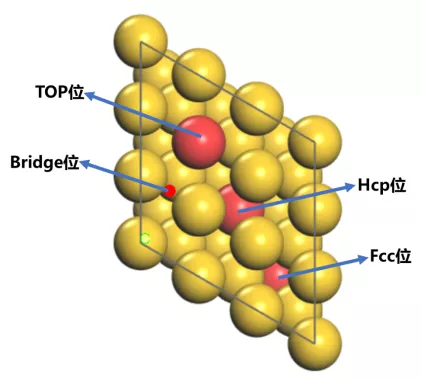
相反,表面的Hollow位正下方(即第二层)有原子,为Hcp位(hexagonal close-packed)。
上述这些吸附位点,在下面这篇文献中做了更加深入和全面的分析,下面的的这张示例图可以作为参考示例,感兴趣的同学可以点击下方详情链接进行查看
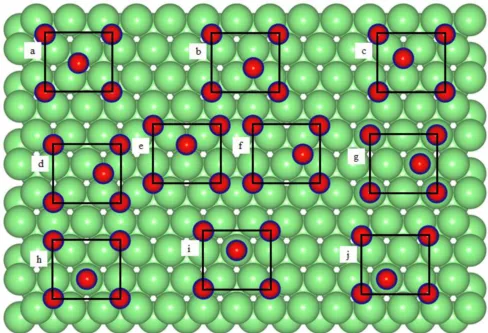
https://www.sciencedirect.com/science/article/pii/S003960281630239
复制蓝色字体下载原文
相信到这里大家已经对具体的吸附位点划分有了详细的了解,现在给大家讲一下怎么来用VASP进行吸附结构优化,这里我们以CO吸附在Au(111)面的TOP位为例进行计算。
接下来可以准备POSCAR,把CO分子吸附在刚才的Au(111)面上,为了减小计算量小编在这里简单地把Au(111)的超胞缩小为2*2*1的了,并且固定了下面两层的Au原子,大家在实际计算中可以根据具体情况操作。
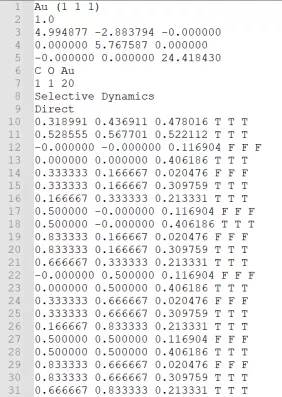
准备好POSCAR后,咱们再着手准备INCAR,KPOINTS和POTCAR
INCAR用到的参数如下:

KPOINTS如下:

POTCAR就不给大家展示了,生成POTCAR的方法还有很多,大家可以自行进行操作。
现在我们已经准备好所有的输入文件,然后选择提交任务就可以进行计算。
小编这里只是以TOP位点作为例子梳理了大概的计算流程,如果有感兴趣的小伙伴可以尝试计算一下CO在其他位点上的吸附结构。
本文所有内容文字、图片和音视频资料,版权均属科学指南针网站所有,任何媒体、网站或个人未经本网协议授权不得以链接、转贴、截图等任何方式转载。









