







 您已经拒绝加入团体
您已经拒绝加入团体

 2021-11-11
2021-11-11
 4549
4549
 0
0
【摘要】 双电层电容器是超级电容器的一个大分支。因为其工作原理相对于赝电容电容器简单,在目前理论研究工作中也相比于赝电容获得了更深入的研究。
【本部分内容简介】
双电层电容器是超级电容器的一个大分支。因为其工作原理相对于赝电容电容器简单,在目前理论研究工作中也相比于赝电容获得了更深入的研究。在本文中,笔者将先从经典的Kornyshev模型出发,介绍如何从简单的晶格气(lattice-gas)模型与理想界面的泊松方程导出在不同电解质条件下的微分电容的解析解。然后介绍几种常用的求解双电层结构和微分电容的模拟方法,包括经典密度泛函理论(Classical Density Functional Theory),巨正则系综蒙特卡洛模拟(Grand Canonical Monte Carlo),经典分子动力学(Classical Molecular Dynamics)。接下来笔者将进一步介绍如何引入恒电压条件与简单扩散动力学到上述方法中以提高对实际体系模拟契合度。以上这些方法被统称为“经典方法”。
经典方法的核心思想是通过热力学与静电学定律,精确求解平衡状态下的双电层的结构(即电解质中离子在电极表面附近分布方式)。经典方法的最大优点是在介观或微观尺度下,可以精确地解得双电层结构与微分电容。电解质的属性,包括阴阳离子的尺寸、价态,浓度等对于经典方法是作为一系列的输入参数来影响双电层的结构的。而电极部分则被当作边界条件或者带电刚体(形状在任何条件下不发生改变)处理。由于经典方法未考虑电极材料的电子结构性质,仅从电解质离子的电吸附响应求算理论电容,某些由电极自身性质贡献出的电容将被完全忽略。例如石墨烯或类石墨烯材料的独特电子结构与介电屏蔽效应所产生的电容便不能被经典方法考虑到。
因此,在经典方法基础之上,笔者将介绍两种主流考虑电极性质的模拟方法:量子-经典结合法和量子力学-溶剂化模型方法。此两种方法也各有特点,在后文中笔者讲详细介绍这些方法的原理,差别,适用情况,局限性,以及相关代表性工作。为了避免繁复的数学推导与过高的专业性,笔者在后文将仅介绍各种方法的基本原理、特点与最重要的结论。对相应技术细节感兴趣的读者可以参考文后的参考文献。
【正文】
一、Kornyshev模型:基于界面泊松方程与晶格气模型的双电层模型
当金属电极与电解质接触后,该电极-电解质系统的电容主要来源于金属-电解质界面的双电层。Gouy-Chapman-Stern (GCS)模型是在众多经典的电化学模型中(应用最广的一种描述双电层结构的方法。如图1所示,GCS模型综合了Helmholtz模型和Gouy-Chapman模型,认为该双电层包括两部分:靠近电极表面的紧密层(compact layer)与距离电极表面稍远的扩散层(diffuse layer)。GCS模型认为,体系的静电势在紧密层线性变化,然后在扩散层呈指数变化。借助德拜-休克尔(Debye-Hückel)近似[注释1],通过一些数学推导可得到体系的静电势分布,并且得到体系的微分电容(differential capacitance,Cdiff)的解析解[公式(1)和(2)]。在这个模型中,一个重要的结论是:微分电容在零电荷电位处(即电极表面电荷密度为零)最小。这是因为sinh 函数在零点处的斜率最小。

这里σ表示电极表面电荷密度,Ψ为静电势,z为阴阳离子带电荷量(这里认为阴阳离子带电量相等,即z+ = z- =z),kB为玻尔兹曼常数,T为热力学温度,f(σ)为表面电荷校正项。
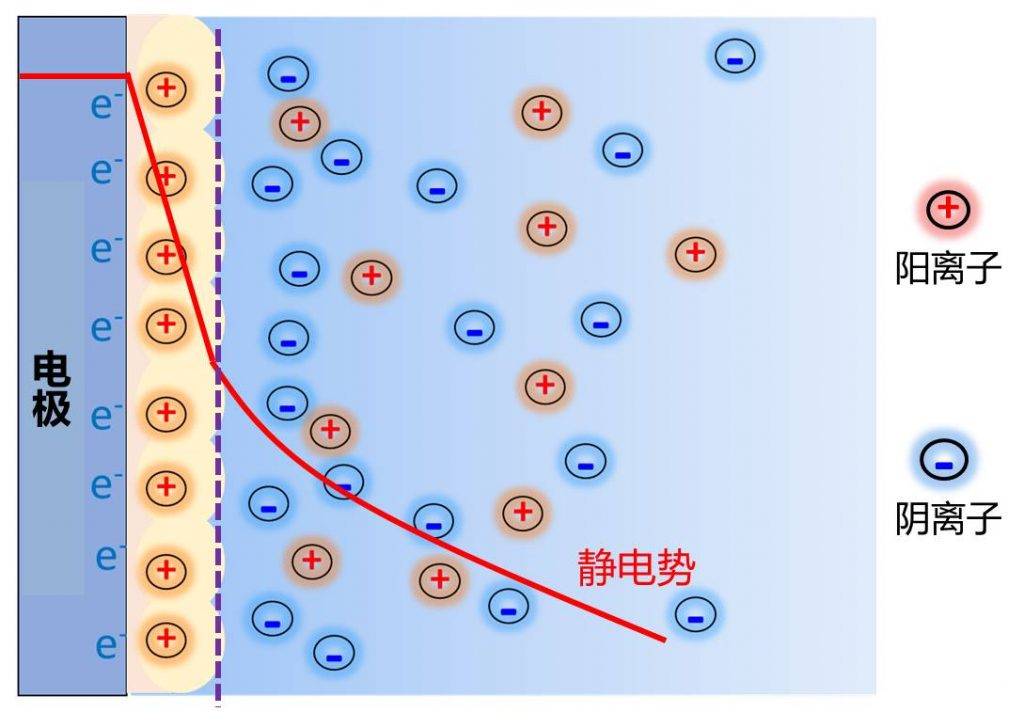
图1. GCS双电层模型示意图。图片来自作者。紫色虚线以内为紧密层,以外为扩散层。
随着材料科学的发展,离子液体作为一种新型的电解质开始进入科研者的视野。离子液体是一类室温下呈液态的离子盐。因其能提供比水性电解质更大的电化学窗口而变得愈发火热(编者注:超级电容器的电化学窗口正相关于器件能够存储的总电量)。然而,实测的金属-离子液体体系的微分电容与GCS预测结果相悖。这就说明GCS这种传统的电化学模型对于离子液体电解质是无效的。实际上,这一结果可以从理论上预料到:因为德拜-休克尔近似只适用于稀溶液。而离子液体仅由阴阳离子组成,它的空间电荷密度(也可理解为离子的体积摩尔浓度)远大于水溶液电解质。这种极高浓度环境下阴阳离子间的排斥作用将比稀溶液中明显很多,因而德拜-休克尔近似的前提无法满足。由此推之,研究离子液体与金属界面的双电层结构与电容行为必需新的双电层模型。
Kornyshev教授在2007年的一篇发表在J. Phys. Chem. B上的feature article中给出一个可能的模型。1(作者注:在应用数学与物理领域的一些更早的文献里可能已经出现了下面将要介绍的模型和推导,但鉴于Kornyshev教授是理论电化学与电容模拟领域的执牛耳者,因此这里选择他的工作介绍。)Kornyshev通过结合平均场理论,晶格气模型(原文称为mean-field lattice-gas model,其基本思想是把离子当做带电粒子放入固定格点中,具体的推导和原理见参考文献2)与界面泊松方程,推导出了一个描述体系静电势分布的方程(在原文中定义为“Poisson-Fermi equation”),然后进一步推导出微分电容的解析解,如公式(3)所示(具体数学推导过程请参见文献1):
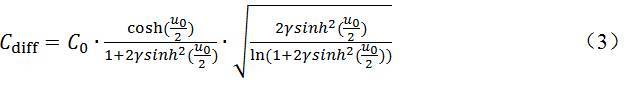
这里C0为一个常数(与溶液浓度,介电常数等属性有关),u0为约化表面电势,γ为约化浓度(此处“约化”指无量纲,是为了便于方程推导而使用的)。在低浓度极限下(γ→0),公式(3)可转化为GCS模型方程。从图1(a)和图1(b)中可以清楚地看出浓度对体系静电势的影响以及晶格气模型相对于GCS模型的偏离程度。当表面电势比较低时[图2(a)],电解质浓度对体系静电势的影响并不大。而当表面电势较高时[图2(b)],浓度对静电势则有很明显影响:低浓度下[注释2],从左向右(即从电极表面向电解质主体),静电势在靠近电极表面处急剧下降;在经过拐点后,开始平缓下降。而在高浓度下,静电势的下降速率在整体上更加平缓。这一现象并不难理解:高斯定理表明电势的二阶导数是空间电荷密度。图2(b)中的拐点前后斜率突变。因而可以将拐点处视作一层较薄的电荷层。该电荷层可对应GSC模型中的紧密层。这与如前所述的结论“低浓度下的实际双电层结构趋近于GCS模型”一致。当浓度增大时,电势的下降则依赖于电解质内部整体离子所决定,而不仅仅与靠近电极表面的部分相关。这也就是说,由于在高浓度条件下的离子关联性(例如空间排斥效应等)增强,此时紧密层与扩散层的边界变得模糊。因此,为了从理论模拟中了解双电层储电的机理与固液界面离子的分布方式(即双电层的结构),我们需要一些计算方法来获得更为精细的双电层结构(后文中将有详细介绍,这里暂不展开介绍)。将公式(3)作图可得到微分电容与电压关系曲线,如图2(c)所示。Kornyshev模型给出了一个适用于金属表面-离子液体微分电容的一般性描述:微分电容-电压曲线在低浓度下呈现驼峰型(camel shape,双峰),高浓度下呈现铃铛型(bell shape,单峰)。当改变阴阳离子尺寸时,峰形和峰位置均会变化[图2(d)]。
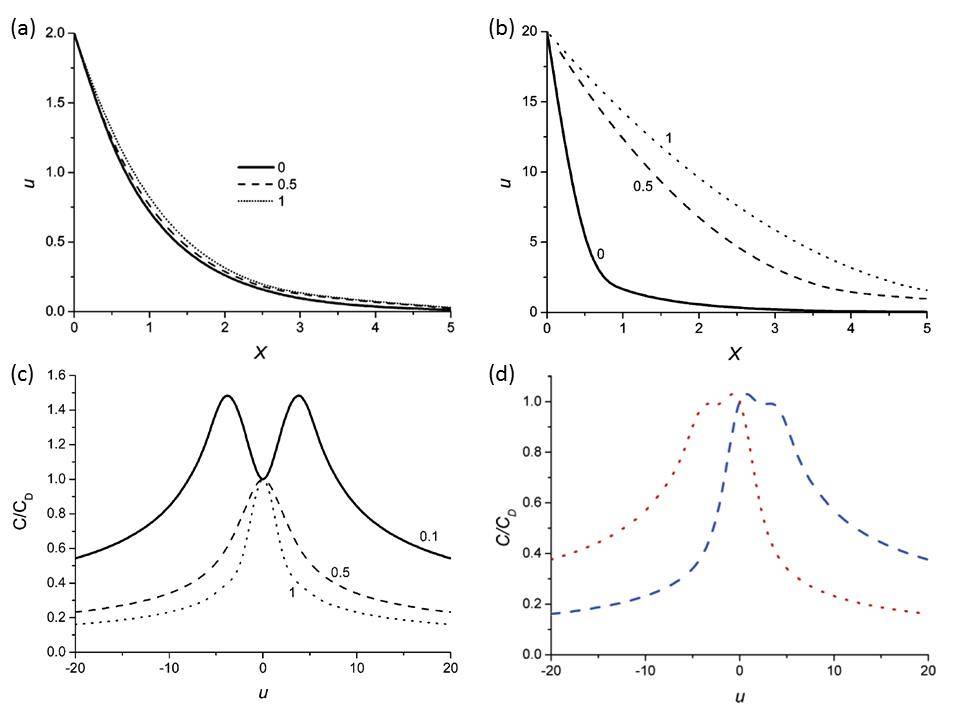
图2. (a) 约化表面电势为2的情况下,不同电解质浓度下的体系静电势分布。纵轴为静电势大小,横轴为到电极表面的距离。0代表电极表面所在的位置。图(b)同。(b)约化表面电势为20的情况下,不同电解质浓度下的体系静电势分布。(c)约化表面电势为20时,不同浓度下体系的微分电容与静电势的关系。(d) 阴阳离子大小不对称的情况下(红点线:阴离子大于阳离子;蓝点线:阳离子大于阴离子),体系的微分电容与静电势的关系。图片均来自于参考文献1。
二、经典密度泛函理论:基于结构热力学与粗粒化模型的双电层求解
经典密度泛函理论(Classic Density Functional Theory, CDFT)是一种适用于介观和微观体系的结构热力学模拟方法,过去往往被用于模拟介孔微孔材料的气体吸附以及胶体大分子的热力学行为。其核心思想是将体系的自由能表达为密度泛函,通过寻找最小自由能来求解体系中的粒子在热力学平衡态下对应的密度分布,即“结构”( CDFT中所模拟的粒子并不是一个具体的原子或分子,而是刚性球粒子,属于粗粒化模型)。在此理论上,如果把粒子之间的静电作用和粒子-外电场相互作用考虑到自由能泛函中,则这一方法便可用于求解前文探讨的金属电极-离子液体的双电层结构。在固定电极表面电荷密度的情况下,可以解出阴阳离子在平衡态下的密度分布。最后通过高斯定理得到体系的静电势分布与电极表面电势的关系(具体的理论推导部分见参考文献3, 4)。从图3(a)和图3(b)中可以看出,离子液体的浓度对电极表面电荷密度-电极表面电压曲线,微分电容都有着很重要的影响。此外,CDFT可以给出更具体的双电层的微观结构。例如从图3(c)中可以看出,低浓度离子液体对表面电压升高的响应主要集中在靠近电极表面处,且只有一层。远离电极表面处,离子分布密度不随电压变化。而对于高浓度的离子液体,电极表面电荷对阴阳离子分布密度的响应则可以延续到更远处:阴离子与阳离子分布密度呈现震荡衰减[图3(d)]。这就是理想离子液体模型(即同尺寸反电荷刚性球模型)的精细双电层结构。CDFT还可通过改变离子尺寸,浓度,价态等参数,甚至加入显性或隐性的溶剂化模型来模拟更加真实的离子液体-金属电极双电层结构,具体可参考目前已发表的一些工作。5-8
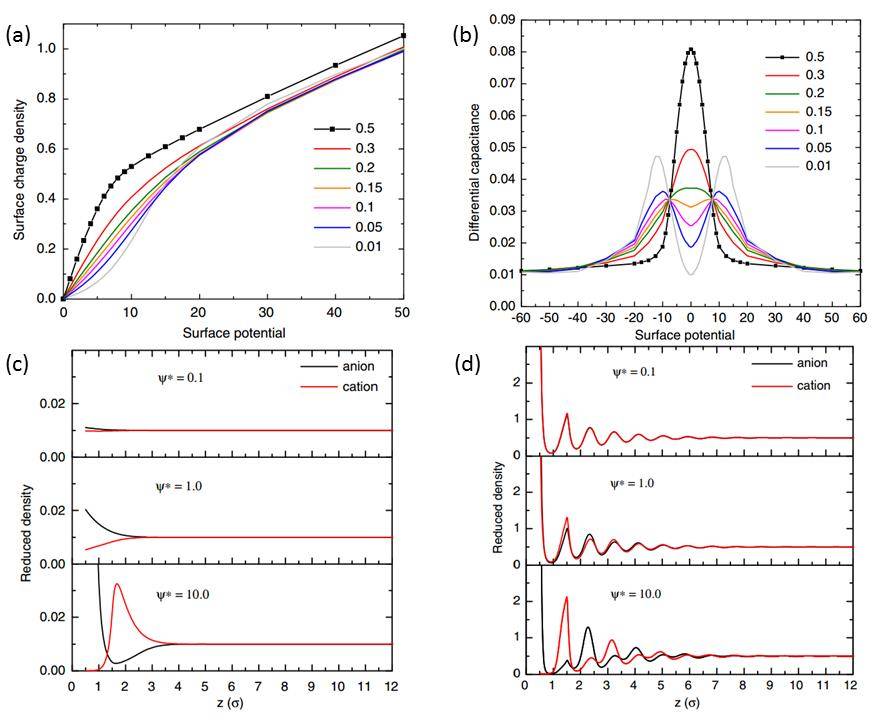
图3. (a)不同浓度离子液体的表面电荷-电压曲线;(b)不同浓度离子液体的微分电容-电压曲线;[编者注:a图中的函数求一阶导数便转化为b图。](c)低浓度离子液体在不同电压下的界面离子分布;(d)高浓度离子液体在不同电压下的界面离子浓度分布。图片来自参考文献4。
三、经典分子动力学与巨正则蒙特卡洛:另外两种求解双电层结构的方法
求解双电层结构实际上就是求解一个体系在有外电场作用下的热力学平衡态结构。对于获得热力学平衡态,其方法并不只局限于前文介绍的经典密度泛函方法,CMD与GCMC也可以得到等效的结果。关于经典分子动力学(Classic Molecular Dynamics, CMD)和巨正则蒙特卡洛(Grand Canonical Monte Carlo, GCMC)的基本理论,因为常见的计算物理教材中均有很详细的介绍,笔者在此不做赘述。
在现阶段,常用的描述离子液体的参数已经优化得非常好了,基本能够比较好地重现其物理化学性质,例如粘度,密度,扩散系数等等。在使用CMD来模拟界面双电层时,只需要手动修改电极表面原子的力场参数,即原子上的电荷量(partial charge)。然后在系统达到平衡态后,统计离子分布并用高斯定理推导出电势分布,即可推算出单位表面面积的电容大小。基于全原子模型的CMD方法的最大优点就是其模拟的离子液体体系十分贴近实际表征(原子力显微镜,中子散射等)结果:离子的空间位阻和离子间的作用(范德华作用,静电作用等)都精确地包含在了用于模拟的力场参数中。其缺点则是模拟的结果可能会非常依赖于力场参数。由于电解质分子自身可能在外加电场下发生极化,因此在某些CMD模拟中可能需要引入极化力场来得出更为准确的双电层结构。另外,在CMD中,通过调整电极每个原子上的电荷来进行充电模拟这一做法在电极表面结构较为复杂时可能会不适用。这是因为当表面结构较为复杂时,如何将电荷分配到不同位置上的原子将会变得棘手。引入恒电压条件,允许原子电荷浮动可解决这一问题(后文将介绍恒电压的引入方法)。关于CMD在固液界面与双电层模拟中的应用的更多细节,可以参考Bedrov教授在2015年发表在J. Phys. Chem. Lett. 上的一篇综述。9
GCMC模拟作为一种常用的热力学模拟手段,在经过改进后也可以用于固液界面的双电层结构求解。在模拟离子液体时,可以直接用刚性带电球来代表阴阳离子。而对于电极模型,则可以使用理想单板或者双板模型,甚至直接对多孔碳的原子模型作GCMC吸附模拟以确定模型。和CMD相比,GCMC方法更加快速,但在双电层结构的细节上不如前者。另外,在GCMC模拟中加入显性的溶剂分子(即在模拟中引入粒子作为溶剂分子)可能会引发许多问题,例如溶剂和离子的浓度相差很大时,对溶剂分子和离子的随机操作频率将很难合理设定。所以采用GCMC来模拟界面双电层时,需要避免使用显性溶剂化模型。近年的发表的一些采用GCMC模拟方法的典型工作请见参考文献。10, 11
四、恒电压条件与扩散动力学的引入,以及几种经典方法的优劣
在CMD模拟中,通过改变电极上的原子电荷来进行充电和双电层模拟是一种常用且快捷的方法。但此方法的一个缺陷是没有考虑到金属电极是等势体(编者注:表面各处电势相等的物体)这一事实,因此在物理上有瑕疵。为了弥补这一不足,可使用恒电压方法改良CMD法。恒电压CMD的基本思想是固定体系中的总电荷量,固定电极表面处静电势,但允许电极上的原子的电荷有浮动。原子电荷采用高斯分布来描述,其展宽为一可调参数。目前已经有许多文献报道了用恒电压方法(固定表面电压优化双电层结构)模拟双电层结构并和CMD传统的恒电荷方法(即固定表面电荷优化双电层结构)模拟结果进行比较。基本结论是:在低电压时,恒电压与恒电荷CMD给出的双电层结构与电容预测相互差别不大。而在高电压时(如大于3 V),这两种方法所预测的双电层结构出现明显差别(具体原因分析可见参考文献12)。在CDFT中,表面电势可以作为泊松方程的边界条件而取一个定值,并在固定表面电势(边界条件)的情况下,优化体系的自由能,同时解出泊松方程。由于CDFT是基于一维结构的模型(电势是一个只与电极表面距离有关的函数),且在数学上十分严谨,因此恒电压法和恒电荷法可以得到完全一样的电荷-电压曲线,在理论上完全自洽。
对于GCMC模拟,由于其可以直接模拟两电极体系,因而固定电极电势的方法是设定一个恒定的两极间电压差(此电压差实际上是体系热力学自由能中的一个热力学参数,跟温度,压强等物理量一样。)通过对离子进行随机操作,可以获得电极上的平均电荷密度的函数,再乘以电压差得到能量函数。具体操作可参阅文献。10
在超级电容器的理论研究领域,离子动力学也是一个十分重要的因素。动力学方面的研究主题主要有两方面:一是离子在带电介孔与微孔[注释3]中的扩散能力,这关系到电容器在快速充/放电时所能存储/提供的电量大小;另一方面则是电极电势随时间改变时,相应的离子的运动图像与双电层的结构变化。前者可以采用CMD进行模拟,直接算出平衡态下离子在带电孔中的扩散系数,此方面已有相关的工作报道。13 例如图4中表明,离子液体中的离子在微孔中的扩散系数要低于离子在纯离子液体自身中扩散系数,且扩散系数还与微孔所带的电荷有关联。电荷与扩散系数的关系需要运用宏观的流体运动理论,例如菲克定律,Navier–Stokes 方程等来阐明。近几年已有一些工作将CDFT与流体力学相结合,发现了不少有趣的物理现象。14-16
前面介绍了三种经典方法:CDFT,CMD,以及GCMC。这里我们做一个简单小结。CMD的优势在于能够模拟真实的溶剂分子和离子液体,在分子原子尺度下给出精细的双电层结构。其缺点则是大体系(即分子和原子个数多)的计算成本高。使用粗粒化的模型(coarse-grained CMD)可减小计算量,这里不再展开介绍。此外,在CMD中引入恒电压条件也将大大增加计算成本。CDFT的优点是运算速率快,且计算成本与体系的大小无关。CDFT是基于严谨的数学表达的模拟,因此它可以给出比较清晰的物理图像和理解。此外,CDFT还可以与流体力学相结合。CDFT的缺点是受数学的局限性很大。对于一维体系(离子密度只在一个维度上与位置有关),CDFT可以解决。但是当离子密度与两个或两个以上的位置参数关联时,CDFT将面临很困难的泊松方程数值求解问题。GCMC的计算成本基本介于CMD与CDFT之间。其优点是可以在恒电压条件下直接模拟两电极体系,对电极结构也没有严格限制。缺点是难以加入溶剂分子,且无法模拟任何动力学信息。

图 4. 离子液体在带电孔中的扩散系数。图片来自参考文献13。
五、考虑电极材料自身性质对电容之贡献:量子电容的引入与溶剂化模型
我们之前所介绍的几种模拟双电层的方法(即经典模拟方法)都是从热力学与静电学的角度出发的。经典模拟方法可以有效地解决复杂体系中的双电层结构问题,但是却忽略了电极材料本身对于电容的影响。近几年来,随着新型电极材料的发展,尤其是二维材料的兴盛,人们发现电极材料自身的性质也会对超级电容器体系贡献电容。这一现象的最典型例子是石墨烯(或类石墨烯)电极体系的量子电容(Quantum Capacitance , CQ)。CQ源于材料自身在费米能级附近的电子能级密度(DOS):当电极材料的DOS比较小时,充放电造成的费米能级移动将会贡献额外的电压变化,导致总电容降低(编者注:电容反比于每充入或放出单位电量的电荷时,电极电势的变化大小);反之,当电极材料的DOS很大时,充放电造成的费米能级移动只会贡献很小的电势变化(图5)。此时电极可视作传统的金属电极,并且可用经典模拟来进行求解,忽略CQ带来的影响。因此,对于石墨烯电极,体系的总电容应贡献自两部分:电极的CQ与电解质的双电层电容(CEDL)。CQ可以用量子力学(如Kohn-Sham密度泛函理论)求出。CEDL则可用前述经典模拟方法求解。最后合二为一即可求出总电容。采用这种方法的模拟就是量子-经典结合法,最早由Hwang教授提出(如图6所示)。17这个模型对理解石墨烯电极的电容行为有着很重大的意义。Ruoff教授于2011年发表在Energy Environ. Sci.上的一篇论文对单层石墨烯的电容有了非常好的阐释,并且发现理论计算给出的石墨烯CQ与电解质CEDL相加合得到的总电容,几乎与实测值完美吻合。18 此外,CQ的引入也可以合理地解释杂原子(如氮)掺杂的石墨烯和有拓扑缺陷的石墨烯具备比纯石墨烯更高的电容(编者注:杂原子掺杂和拓扑缺陷的存在会改变石墨烯DOS值)。

图5. 量子电容与DOS的简单阐释。(a)当DOS很大时,电极上的电子得失对费米能级的位置(图中灰色区域和白色区域交界处)影响很小(深灰色区域为充电前电极材料能级结构,浅灰色为充入电子后电极材料整体系增加的能量,b同)。(b)当DOS较小时,电子得失会导致费米能级位置发生显著变化。图片来自作者。
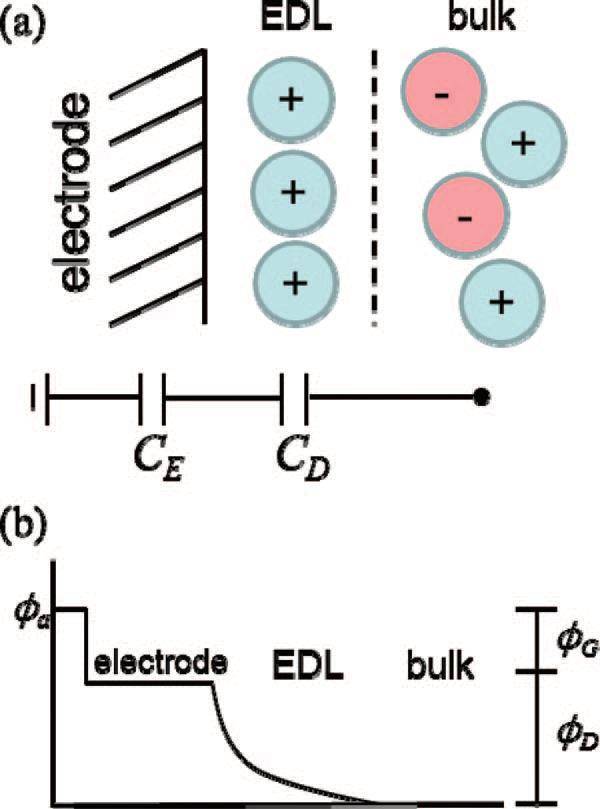
图6. (a)量子-经典结合法模型的示意图,电极为石墨烯,EDL表示双电层。(b)量子-经典模型的电势变化贡献示意图。CE为电极自身的电容,即量子电容。图片来自参考文献17。
除了量子-经典结合法,也可以直接通过结合量子力学与连续溶剂化模型来求解电极的总电容。这里的溶剂化模型并非一般的计算化学中使用的溶剂化模型(如PCM,COSMO等),而是指一些适用于电化学模拟的特殊的溶剂化模型。采用这种模型的方法被称为量子力学-溶剂化模型方法。在这里简单介绍两种模型:ESM(Effective Screening Medium19)和线性 PCM(Linear Polarizable Continuum Model20)。
ESM的核心做法是在电极上面的真空层中填入电介质,并且当电极上有净电荷时,在电介质中给出一层符号相反的镜像电荷,以此来模拟电解质中的紧密层。而线性PCM则是在传统的PCM中,加入线性响应近似(linear response approximation)和非均相泊松玻尔兹曼方程(inhomogeneous Poisson-Boltzmann equation)来模拟电解质对电极上的充放电行为的响应。在这两种模型中求解总电容的方法是一样的:通过计算电极的电化学势随电极上净电荷的变化,得出对应的电荷-电压关系以及微分电容。而体系的量子电容,则可以通过将费米能级变化从电化学势变化中分离出来而得到。(关于理论电容的贡献分解与内部物理机制,笔者将会在后面部分的系列文章中详细阐述。)
量子-经典结合法的优点是在引入量子电容后,精细的双电层结构仍然可以被描述出来,因此该法可应用于各种电解质体系(水溶液和离子液体皆可)与规整的二维材料电极。其缺点是因为CQ与CEDL分别由量子力学与经典模拟求出,忽略了电极与电解质在微观层面上的相互作用,物理上不自洽。其次,当电极材料有一定厚度时,量子-经典方法无法给出电极内部的静电势分布,因此也无法考虑电极自身在双电层电场下的极化与介电屏蔽等效应。量子力学-溶剂化模型方法无法得出双电层精细结构,但电极部分获得的信息则非常详细,可涵盖静电势分布,电荷分布,介电屏蔽贡献等方面。因此,量子-经典方法是以电解质效应为主,电极效应为辅,主要关注两者之间的竞争与支配关系;而量子力学-溶剂化模型则是以电极效应为主,电解质效应为辅,主要关注电极自身的性质。
【注释】
1.德拜-休克尔近似针对电解质溶液做出了下列几个近似:1)对于强电解质,其在溶剂中完全解离为阴阳离子;2)离子为带电刚体;3)阴阳离子间作用力只存在静电库仑力;4)阴阳离子间吸引能小于自身运动动能;5)溶剂为连续介质,决定介电常数。
2.离子液体的“浓度”在模拟计算中是指“空间密度”,不是常规溶液浓度的概念。
3.根据国际纯粹与应用化学联合会(IUPAC)的命名原则,介孔指孔径为2-50 nm的孔隙;微孔指孔径小于2 nm的孔隙。
【参考文献】
(1) Kornyshev, A. A. Double-Layer in Ionic Liquids: Paradigm Change? J. Phys. Chem. B 2007, 111, 5545-5557
(2) Bernard, M. O.; Plapp, M.; Gouyet, J. F. Mean-field kinetic lattice gas model of electrochemical cells. Phys Rev E 2003, 68,
(3) Wu, J. Z.; Li, Z. D. Density-functional theory for complex fluids. Annu. Rev. Phys. Chem. 2007, 58, 85-112
(4) Jiang, D. E.; Meng, D.; Wu, J. Z. Density functional theory for differential capacitance of planar electric double layers in ionic liquids. Chem Phys Lett 2011, 504, 153-158
(5) Henderson, D.; Jiang, D. E.; Jin, Z. H.; Wu, J. Z. Application of Density Functional Theory To Study the Double Layer of an Electrolyte with an Explicit Dimer Model for the Solvent. J. Phys. Chem. B 2012, 116, 11356-11361
(6) Jiang, D. E.; Jin, Z. H.; Henderson, D.; Wu, J. Z. Solvent Effect on the Pore-Size Dependence of an Organic Electrolyte Supercapacitor. J. Phys. Chem. Lett. 2012, 3, 1727-1731
(7) Jiang, D. E.; Wu, J. Z. Microscopic Insights into the Electrochemical Behavior of Nonaqueous Electrolytes in Electric Double-Layer Capacitors. J. Phys. Chem. Lett. 2013, 4, 1260-1267
(8) Jiang, D. E.; Wu, J. Z. Unusual effects of solvent polarity on capacitance for organic electrolytes in a nanoporous electrode. Nanoscale 2014, 6, 5545-5550
(9) Vatamanu, J.; Bedrov, D. Capacitive Energy Storage: Current and Future Challenges. J. Phys. Chem. Lett. 2015, 6, 3594-3609
(10) Kiyohara, K.; Asaka, K. Monte Carlo simulation of electrolytes in the constant voltage ensemble. J. Chem. Phys. 2007, 126, 214704
(11) Kiyohara, K.; Asaka, K. Monte Carlo simulation of porous electrodes in the constant voltage ensemble. J. Phys. Chem. C 2007, 111, 15903-15909
(12) Merlet, C.; Pean, C.; Rotenberg, B.; Madden, P. A.; Simon, P.; Salanne, M. Simulating supercapacitors: can we model electrodes as constant charge surfaces? J Phys. Chem. Lett. 2013, 4, 264-268
(13) He, Y. D.; Huang, J. S.; Sumpter, B. G.; Kornyshev, A. A.; Qiao, R. Dynamic Charge Storage in Ionic Liquids-Filled Nanopores: Insight from a Computational Cyclic Voltammetry Study. J. Phys. Chem. Lett. 2015, 6, 22-30
(14) Jiang, J.; Cao, D. P.; Jiang, D. E.; Wu, J. Z. Time-dependent density functional theory for ion diffusion in electrochemical systems. J. Phys.: Condens. Matter 2014, 26, 284102
(15) Jiang, J.; Cao, D. P.; Jiang, D. E.; Wu, J. Z. Kinetic Charging Inversion in Ionic Liquid Electric Double Layers. J. Phys. Chem. Lett. 2014, 5, 2195-2200
(16) Lian, C.; Gallegos, A.; Liu, H. L.; Wu, J. Z. Non-Scaling Behavior of Electroosmotic Flow in Voltage-Gated Nanopores. Phys. Chem. Chem. Phys. 2017, 19, 450-457
(17) Paek, E.; Pak, A. J.; Hwang, G. S. A Computational Study of the Interfacial Structure and Capacitance of Graphene in [BMIM][PF6] Ionic Liquid. J. Electrochem. Soc. 2013, 160, A1-A10
(18) Stoller, M. D.; Magnuson, C. W.; Zhu, Y. W.; Murali, S.; Suk, J. W.; Piner, R.; Ruoff, R. S. Interfacial capacitance of single layer graphene. Energ Environ. Sci. 2011, 4, 4685-4689
(19) Otani, M.; Sugino, O. First-principles calculations of charged surfaces and interfaces: A plane-wave nonrepeated slab approach. Phys. Rev. B 2006, 73, 115407
(20) Letchworth-Weaver, K.; Arias, T. A. Joint density functional theory of the electrode-electrolyte interface: Application to fixed electrode potentials, interfacial capacitances, and potentials of zero charge. Phys. Rev. B 2012, 86, 075140
科学指南针是互联网+科技服务平台,500多家检测机构,提供近5万种设备和服务项目,涵盖生物医药、智能硬件、化学化工等多个领域,由专业人员1对1跟踪服务,保证检测质量与效率。
免责声明:部分文章整合自网络,因内容庞杂无法联系到全部作者,如有侵权,请联系删除,我们会在第一时间予以答复,万分感谢。









