







 您已经拒绝加入团体
您已经拒绝加入团体

 2021-09-02
2021-09-02
 2870
2870
 0
0
【摘要】 无论是做第一性原理还是分子模拟计算,我们都是为了实现一定的目的。分子模拟可以计算的内容很多,为了实现不同的目的,需要选择合适的流程、系综等,今天为大家介绍一些分子模拟的常见任务流程。
无论是做第一性原理还是分子模拟计算,我们都是为了实现一定的目的。分子模拟可以计算的内容很多,为了实现不同的目的,需要选择合适的流程、系综等,今天为大家介绍一些分子模拟的常见任务流程。
01
平衡态计算
有时候我们需要研究不同温压条件下模拟体系内的局域结构(RDF、CN等)、简单热力学性质(密度、比热等)。
对于平衡态的计算,通常任务流程为:构建合理的初始结构—设定合适的力场参数—Minimization—NVT—NPT—NVT(采样)—数据后处理—绘图。
# Simulation details
units real
dimension 3
boundary p p p
atom_style full
pair_style lj/cut/tip4p/cut 1 2 1 1 0.1546 10 8.5
bond_style harmonic
angle_style harmonic
# 构建合适的初始结构
read_data model.data
# 设定合适的力场参数
pair_coeff 1 1 0.1852 3.1589
pair_coeff 1 2 0 0
pair_coeff 2 2 0 0
bond_coeff 1 527 0.9572
angle_coeff 1 37.95 104.52
group water type 1 2
velocity all create 298 123456 mom yes rot yes dist gaussian units box
thermo 100
thermo_style custom step temp press vol etotal ke pe
# 能量最小化
minimize 1.0e-4 1.0e-6 1000 10000
# NVT
fix 1 water shake 1.0e-4 200 0 b 1 a 1
fix 2 nvt temp 298 298 100
run 50000
unfix 2
# NPT
fix 3 npt temp 298 298 100 iso 10 10 1000
run 50000
unfix 3
# NVT, sampling
fix 4 nvt temp 298 298 100
run 50000
write_data H2O.data
02
非平衡态计算
在一些特殊的情况,需要对体系施加外力(电场、磁场等),使得体系随时间不断变化,我们称之为非平衡态计算。
这里以RNEMD方法计算粘度为例介绍其基本流程:构建合理的初始结构—设定合适的力场参数—Minimization—NVT—NPT—NVE(fix viscosity,采样)—数据后处理—绘图。
图2中1-41行与平衡态计算一致,特别注意44-47行。其中44-45行对模拟体系进行分层,共等分为1/0.05=20层,并记录了每一层x方向速度;46行fix viscosity执行RNEMD算法,详情见链接:
https://docs.lammps.org/fix_viscosity.html;
47行对动力学进行积分,更新原子坐标与速度。计算完成后,从lammps的log文件内获取动量交换量,从velocity_profile.dat文件获取体系速度分布;
根据公式进行计算得出:
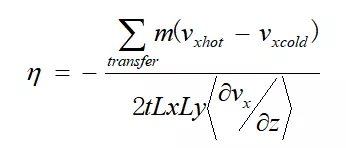
# Simulation details
units real
dimension 3
boundary p p p
atom_style full
pair_style lj/cut/tip4p/cut 1 2 1 1 0.1546 10 8.5
bond_style harmonic
angle_style harmonic
# 构建合适的初始结构
read_data model.data
# 设定合适的力场参数
pair_coeff 1 1 0.1852 3.1589
pair_coeff 1 2 0 0
pair_coeff 2 2 0 0
bond_coeff 1 527 0.9572
angle_coeff 1 37.95 104.52
group water type 1 2
velocity all create 298 123456 mom yes rot yes dist gaussian units box
thermo 100
thermo_style custom step temp press vol etotal ke pe
# 能量最小化
minimize 1.0e-4 1.0e-6 1000 10000
# NVT
fix 1 water shake 1.0e-4 200 0 b 1 a 1
fix 2 nvt temp 298 298 100
run 50000
unfix 2
# NPT
fix 3 npt temp 298 298 100 iso 10 10 1000
run 50000
unfix 3
# NVE, RNEMD方法计算粘度
compute layers all chunk/atom bin/1d z center 0.05 units reduced
fix 4 all ave/chunk 20 50 1000 layers vx file velocity_profile.dat
fix 5 all viscosity 100 x z 20
fix 6 nve
variable dVx equal f_4[11][3]-f_4[1][3]
thermo 1000
thermo_style custom step temp press etotal v_dVx f_5
run 500000
03
更复杂的情况
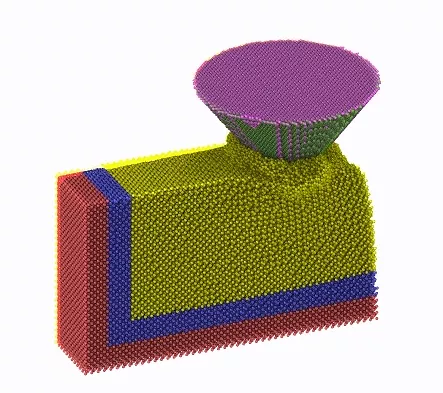
复杂的模拟可以被拆解为几个部分,无非也是选择合适的系综,对体系部分原子进行一定的限制。以圆锥形SiC磨粒磨削单晶Si为例,模拟流程为:
(1)建模:单晶硅分为三个区域:边界区、恒温区、牛顿区。磨粒为圆锥形,材料为SiC。
(2)模拟初始化,固定边界原子,设置磨粒原子为刚性原子。
(3)能量最小化,对系统进行弛豫,使体系能量达到最低。
(4)设置恒温区温度为300K。
(5)设置系综,设置磨粒移动方向和速度。
(6)输出轨迹文件、输出三个方向受力。
以上是常见的分子模拟计算任务流程,具体案例说明见下期分享~









